
全体を通してやりたいこと
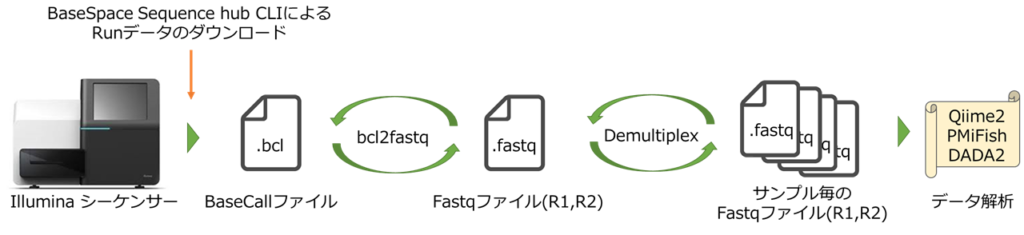
上記図はIlluminaシーケンサーから得られたbclファイルに対して、処理を加えていき最終的にデータ解析に使用するサンプル毎に配列情報が配分されたfastqファイルを作成する順序を示しています。
bclファイルが生成された後の作業を自身のPC内のローカル環境で実行していきたいと思います。
とりあえずPart1では、MiSeqの使用を前提としてオレンジ矢印の部分について説明していきます。また、ついでにProjectごとにFastq.gzをダウンロードする方法についても記載します。
1. なぜ、”ネット経由で”なのか
いくつか動機となる理由がありますが、主なものとしてはリモートワーク中にシーケンスが終了した際、すぐにデータ解析を進めるためです。また、データ処理は最終的になんでもパイプライン化したいので、コマンドベースで指示をまとめられると理想的です。
そのため、データをネット経由で自身の社内PCに移動する方法を模索していました。
2. 一般手的なFastqファイルの取得
Fastqファイルの大元はBaseCallファイルです。IlluminaシーケンサーはサイクルごとにATGCの蛍光を画像で取得しシグナルを抽出することでBaseCallファイルを作成します。
Illuminaシーケンサーのシーケンス原理については、以下の資料が分かりやすいです。

Miseqの場合は配列を決定する時の蛍光取得は、1サイクルごとにATGCの塩基それぞれで写真を撮影し、その写真の情報をもとに1サイクルごとのBinary Base Call (bcl) ファイルが生成されます。
特にプロトコル通りにシーケンスを進めていれば、シーケンサーに搭載されているCASAVAによってbcl2fastqが実行され、.bclファイルから.fastqファイルが生成されます。
この時、サンプルシートに記載したサンプル毎のタグ配列情報をもとにfastqファイルが分割(demultiplex)されて出力されます。
3. ネットワーク経由でRunデータをダウンロード
3-1. ネット経由でデータをダウンロードする方法はどんなものがあるのか
BaseMount
BaseMountはBaseSpace Sequence Hubのデータをマウントするツールらしいです。プロジェクトやサンプル、Runなどの結果を直接操作することもできます。
最初はBaseMountを試しましたが、OpenGPGがないと怒られ、暫く頑張った結果諦めました。
BaseSpace Sequence Hub CLI
BaseSpaceはシーケンスの状況をモニタリングできるIlluminaのサービスで、BaseSpace Sequence Hub CLIはコマンドラインベースのBaseSpaceです。
BaseMountはBaseSpace Sequence Hub APIを使用しているとあったので、今回は大元であろうこちらを利用することにしました(こっちなら使えた)。
3-2. BaseSpace Sequence Hub CLIのインストール
インストールから実行までBaseSpace Developersのページを参考にしています。
必要なものと使用要件は以下の通りです。
- BaseSpaceのアカウント(無料)
- WSL
- Ubuntu 20.04 LTS
BaseSpaceとは?という方はBaseSpace DevelopersのBaseSpace OverView(英語)が分かりやすいです。
環境構築は以下のページの準備 コマンドライン編を参照していただければと思います。

チュートリアル通り、ホームディレクトリにbinフォルダを作成して、wgetコマンドで最新版のファイルをダウンロードします。ここは各自の管理方法に沿って適宜変更してください。
|
1 2 |
# ホームディレクトリに移動 $ cd |
ホームディレクトリにbinフォルダを作成
|
1 |
$ mkdir bin |
binフォルダにダウンロード。-O以降を変更するとインストールする場所を変更できます。
|
1 |
$ wget "https://launch.basespace.illumina.com/CLI/latest/amd64-linux/bs" -O $HOME/bin/bs |
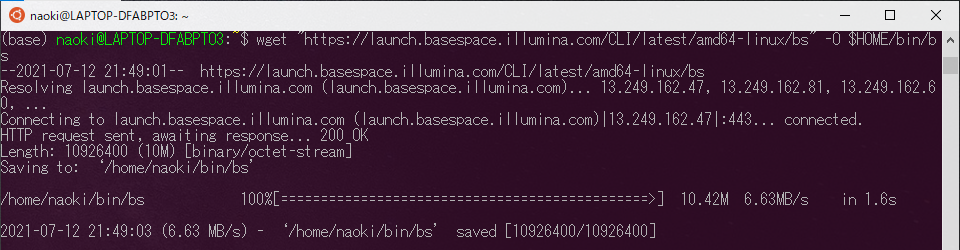
ダウンロードが終わったら、実行権限を与えます。
下記コマンドは、所有者に対して、$HOME/bin/にあるbsを実行する権限を与えています。このコマンドを実行する前にbsを実行すると

アクセスを拒否されます。
|
1 |
$ chmod u+x $HOME/bin/bs |
chmodはファイルやディレクトリに対して様々な権限を設定できるコマンドです。ユーザー区分ごとに権限を設定することができます。
次に、bs コマンドのみで実行できるように、コマンドまでのパスを追記します。
|
1 |
$ export PATH=$PATH:$HOME/bin/ |
上記コードを実行したらhelpオプション-hを付けて実行できるか確かめてみましょう。
|
1 |
$ bs -h |

アカウントの認証
下記コードを実行すると指定したURLに移行するように指示されるので、コピペしてsign inページへ移動します。

いつも通りsign inします。
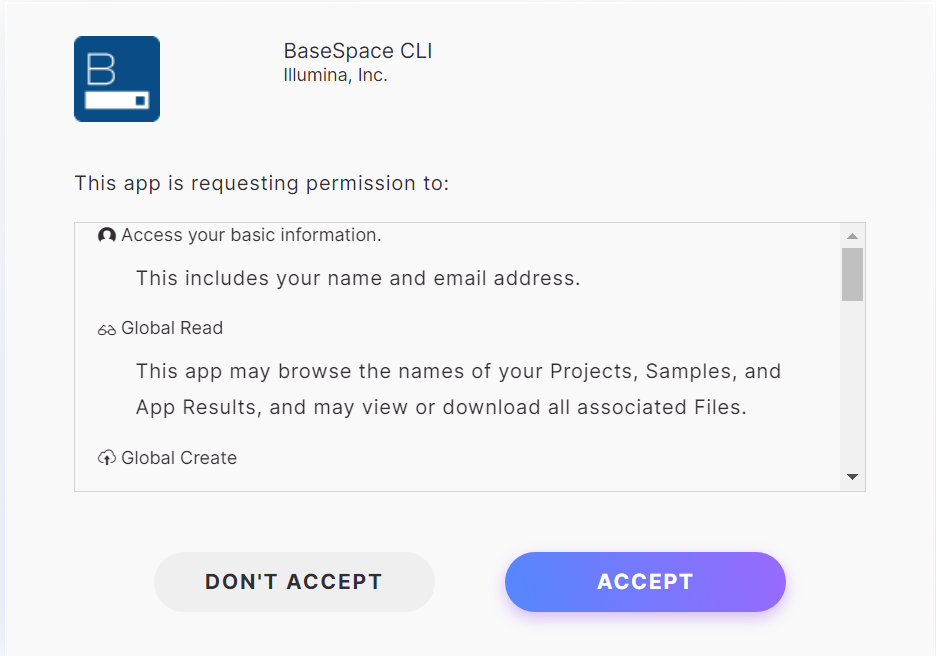
BaseSpace CLIが以下の項目について実行してもいいか聞いてくるので問題なければACCEPTをクリック
OAuthが認証されましたと表示されるので、CLOSEをクリックして終了します。
これでBaseSpace Sequence Hub CLIのコマンドを使ってBaseSpace上のファイルの操作ができるようになりました。
Runデータをダウンロードする(Fastq.gzファイル以外)
RunデータをダウンロードするにはRun IDが必要になります。以下のコマンドを使って簡単に探すことができます。
$ bs list runs
もし上記コードで default.cfgが無い とエラーが出たらOAuthの認証が出来ていないのでもう一度やり直してください。
またはBaseSpaceのRunタブよりダウンロードしたいRunをクリックすると、例えば以下のようなURL構成になっているかと思います。
https://basespace.illumina.com/run/277282/2x151PhiX.
“run/”以降の277282というのが対象のRun IDです。
欲しいRunデータが決まったら以下のコマンドでデータをダウンロードします。
$ bs run download –id=(欲しいRunデータのID) –output ./Rundata
これでGenerateFASTQされる前の状態のoutputファイルが取得できました。
Part2では、ここで得たRunデータフォルダを使用して、bcl2fastqを使ったFastq.gzの生成についてお話しします。
ついでに、 Fastq.gzをプロジェクト単位でダウンロードする
以下のコマンドはSampleSheetのProjectのカラムに記載された情報をもとに、Project単位でFastq.gzをフォルダ分けすることができます。
MiseqのSampleSheetの作成の仕方は下記が参考になります。

(欲しいFastq.gzが所属するProject名) の部分にProject名を記載します。
正規表現が使えるので、例えば、testrun_20211201, testrun_20211202, testrun_20211203というプロジェクトがあった場合には、testrun.*と記載すると上記3つを指定していることになります。
$ bs list datasets –terse -f csv –filter-field=Project.Name –filter-term=(欲しいFastq.gzが所属するProject名) | while read id; do echo ${id}; bs download dataset -i ${id} –extension=fastq.gz -o ./fastqgz/ ;done
これで、Runに関する情報をダウンロードする事が出来ました。
Runが終わったら実行されるようにしておくと自動でFastq.gzフォルダをフォルダ分けしておくことができそうですね。
参考
- Options for downloading run folders from BaseSpace Sequence Hub
- BaseSpace Developers BaseSpace CLI overview, Examples
免責事項
十分注意は払っていますが、本記事の情報・内容について保証されるものではありません。本記事の利用や閲覧によって生じたいかなる損害について責任を負いません。また、本記事の情報は予告なく変更される場合がありますので、ご理解くださいますようお願いします。





