

A novel metabarcoding primer pair for environmental DNA analysis of Cephalopoda (Mollusca) targeting the nuclear 18S rRNA region
Cephalopoda(頭足類)はイカ、タコ、オウムガイなどの貝から足が沢山生えたような生物のことで、その大半はイカとタコが占めています。
日本近海にはイカが120種、タコは50種程度生息しており、特に食用にされるアオリイカやマダコは馴染み深い種です。
頭足類に環境DNAを使用した例だと、 Wada et al.2020のダイオウイカ(Architeuthis dux)の検出などがあります。
ここでは、ユニバーサルプライマーの開発・検討手順に注目しつつ、データベース作成のアプローチについて訳していきたいと思います。
以下はアブストを訳したものです。読み飛ばしてもらっても問題ありません。
この記事の内容
論文要約
頭足類は海洋食物網の中心的な構成要素ですが、多様性の研究は、これらの素早い海洋軟体動物をサンプリングするという課題によって妨げられています。
環境DNAメタバーコーディングは、海洋頭足類の生物多様性と分布を研究するための潜在的に強力な手法ですがこれまで適用されていません。
環境DNAから頭足類をメタバーコーディングするための新しいユニバーサルプライマーペア、Ceph18S(Foward : 5′-CGC GGC GCT ACA TAT TAG AC-3 ‘, Reverse:5’-GCA CTT AAC CGA CCG TCG AC-3’)を提案します。
プライマーペアは核の18S rRNAのV2領域を対象とし、分解されたDNAの増幅を可能にするために約200bpと比較的短い配列を増幅します。
配列が登録されている参照データベースと頭足類の組織から抽出したDNAサンプルでのテストでは、Ceph18Sを使用して、頭足類の約310〜460種に相当する44〜66%を増幅および識別できると推定しています。
以前に発表されている頭足類のミトコンドリア16S rRNAをターゲットとする2つのプライマーセット(Jarman et al. 2006 Mol. Ecol. ; Peters et al. 2015 Mar. Ecol.)とCeph18Sを合わせた複数マーカーによるアプローチは、全頭足類の89%を増幅することができ、また、そのうちの19%はCeph18Sによってのみ同定できると推定されています。
Ceph18Sで取得されたすべてのシーケンスはGenBankに登録したことで、新規的に13の頭足類の18SrRNA配列が得られました。
Material and methods
プライマー作成の方針
メタバーコーディング用プライマーペアの開発は、
1) リファレンスデータベースの構築
2) プライマーセットの特定
3) in silicoによるテスト
4) 実験によるプライマーテスト
の4ステップで進められました。
Step1. リファレンスデータベースの作成
データベースの作成は以下のようなイメージです。NCBIが提供するBLAST検索とrRNAのデータベースであるSILVAを使って対象領域である18s rRNA(核内の領域)の配列取得を行っています。

取得した配列はPythonで稼働するOBITools v.1.2.10を使用して、ecoPCR v.0.2のデータベース形式に変換しました。
Step2. プライマーセットの特定
Step1でGenBankとSILVAから作成したリファレンスデータベースをもとに、OBITools v.1.2.10に実装されているecoPrimer v.0.3を使って種レベルで識別が十分可能な変異を持つ領域を増幅するプライマーを特定しています。
基本的な設定値はTable.1に示した通りです。SILVAのデータベースに適用した際、GenBankから抽出したデータに適用した設定では候補となるプライマーセットを返さなかっため、少し緩めの設定でプライマーの検索をかけています。
| パラーメーター | 変数 | 備考 |
| プライマーの長さ (bp) | 20 | 必要とされるForward, Reverseプライマーの長さ |
| 対象アンプリコンの長さ(bp) | 50-200 | 必要とされるアンプリコンの配列の範囲 |
| strict matching quorum | GenBank: 0.7 SILVA: 0.5 | プライマーと対象配列が完全に一致する参照データベース内の配列数の最小割合 |
| sensitivity quorum | GenBank: 0.9 SILVA: 0.7 | 指定されたパラメーターと完全に一致する参照データベース内の配列数の最小の割合 |
| ミスマッチ数 | GenBank: 0 and 3 SILVA: 3 | 許容されるプライマーと対象配列とのミスマッチ数 |
| 3’末端のマッチ数 | GenBank: NA and 2 SILVA: 2 | 3’末端で必要とされる厳密な一致数 |
GenBankのデータベースを対象とした際に候補となったプライマーは、
- 最低のTm値(プライマーがターゲットの配列と50%の割合で2本鎖を形成する温度)は59-69℃
- プライマー間のTm値の差が3℃未満
- GとCの塩基配列の割合が50-60%
- Gまたは/およびCが4つ以下のGCクランプ
- 4塩基以下のリピート配列
- 2塩基のリピートが4つ以下
でフィルタリングされていました。
SILVAのデータベースを対象とした場合は少し緩和した条件でプライマーの候補を抽出しています。
よって、in silicoテストでBc(データベース内の配列に対して増幅できた分類群の割合)がGenBank由来のプライマーセットと同定以上の候補のみを検討し、Tm値が45-70℃でGCの割合が45-65%であるかどうかでフィルタリングしています。
Step3. in silicoによるテスト
Step2で提案されたプライマーセットから、Bc(データベース内の配列に対して増幅できた分類群の割合)とBs(データベース内の配列に対して種を同定できた割合)が最も高かった8つのプライマーセットを選択しています。
その中から、オンラインのOligonucleotide Properties Calculator v. 3.27を用いて2次構造(ヘアピンとプライマーダイマー)の確認し、オンラインのPrimer BLASTで自己相補性(Self complementarity)とin silicoによる増幅の確認を行いました。
プライマーセットのBcおよびBsはミスマッチを許容せず、SILVAデータベースに対するecoPCR v.0.2を用いたin silicoテストで算出しています。
こうして、2次構造を生成しない or 限定的な場合にのみ生成する、BcとBsの指標が最も高い3つのプライマーセットが選択されました。
これらはStep4の実験によるプライマーテストに使用しています。
既存のプライマーセットとの相性の確認
16s rRNA領域を対象とした以下の既存の2つのプライマーセットと、新たに開発したプライマーセットで結合してしまわないか、相補性を確認しています。
| プライマー名 | 配列 5′ – 3′ | 産物長 | 出典 |
| CephMLSf1 | TGCGGTATTWTAACTGTACT | 70–73 bp | Peters et al. 2015 |
| CephMLSr1 | TTATTCCTTRATCACCC | ||
| S_Cephelapoda- F | GCTRGAATGAATGGTTTGAC | 212–244 bp | Jarman et al.2005 |
| S_Cephalopoda-R | TCAWTAGGGTCTTCTCGTCC |
- blastnの検索プロトコルを使用
- 検索データベースをヌクレオチドデータベースの頭足類分類群に限定
- 未培養/環境サンプルを除外
- クエリのカバー率は50%以上
16s rRNA用のデータベースはそれぞれのプライマーをPrimer BLASTにかけて最初にヒットした配列を、以下の条件でBLAST検索にかけました。
こうして作成された16s rRNAのデータベースを用いて、ミスマッチは許容せずにecoPCR v.0.2によるin silicoのテストを通して、BsとBcを算出しました。
Step4. 実験によるプライマーテスト
それぞれ異なる科の3種類(Bathyteuthis abyssicola, Heteroteuthis dispar, Liocranchia reinhardtii)の組織片を用いたプライマーテストを行っています。
エタノール保存されたサンプルをDNeasy Blood and Tissue KitでDNA抽出しました。
スクリーニング用のPCRのテンプレートとして、
- それぞれを10ng/ulに調整したもの
- 3種を混ぜ合わせたもの
- PCRブランク
を使用して増幅の確認を行っています。
PCRは以下の試薬構成と温度サイクルで行っています。
| 試薬名 | 終濃度 or 添加量 |
| 1×Fidelity バッファー(MgCl2) | 0.4mM |
| KAPA Hotstart ポリメラーゼ | 0.02 U µl−1 |
| Forward Primer, Reverse Primer (終濃度 0.5µM) | 0.5uM each |
| DMSO | 5% |
| KAPA dNTPs | 0.3mM |
| テンプレートDNA | 10uL |
| Total | 40uL |
| PCRステップ | 温度 : 時間 | サイクル |
| DNAの初期変性 | 95℃ : 5min | 1 |
| DNAの変性 (2本鎖→1本鎖) | 98℃ : 20s | – |
| プライマーのアニーリング (Gradient PCR) | 最低Tm値-3℃から3℃ずつ5段階で上昇 : 15s | 35 |
| 伸長反応 (1本鎖→2本鎖) | 72℃ : 1min | – |
| 最終伸長反応 | 72℃ : 10min | 1 |
| Hold | 4℃ : ∞ | 1 |
このスクリーニングテストで最適なアニーリング温度を決定した後、さらに多くの組織片を使って対象領域の配列を増幅して、サンガーシーケンスによって配列決定してデータベースに反映させました。
Results
プライマーペアについて
18sを対象に新規開発した3つのプライマーのうち最もクリーンな増幅が見られたプライマーペアを”Ceph18S”と名付けてメタバーコーディングに使用しました。
Table.2のBcは in silicoのテストで増幅が確認された割合(増幅できた分類群/ 対象分類群)で、BsはSILVAデータベースに基づくin silicoのテストで同定できた種の割合(同定できた分類群 / 増幅できた分類群)を表しています。
| 名前 | Ceph18S |
|---|---|
| Forward | 5′-CGCGGCGCTACATATTAGAC-3′ |
| Reverse | 5′-GCACTTAACCGACCGTCGAC-3′ |
| Forward Tma | 59.3°C |
| Reverse Tma | 61.7°C |
| Target length | ∼150–190 bp |
| Bc | 0.85 |
| Bs | 0.78 |
Ceph18Sは18S rRNA遺伝子のV2可変領域を対象としています(Fig.1)。Fig.1の縦軸は塩基の多様性です。この値が高いと配列がばらばら(=属, 種, 種内で配列が異なる)で、低いと同じ配列を持つ(=保存的領域でユニバーサルプライマーを作成するのに適している)ということです。

Ceph18Sが増幅すると思われる領域を見てみると、(Fig.2a)ecoPCRの対象配列と(Fig.2b)実験によって得られた配列それぞれで、保存された領域と可変性の高い内部領域がしっかり分かれていることが分かります。

Ceph18Sの解像度について
Ceph18Sが頭足類がどの程度の同定能力を持っているかについてです。
SILVAデータベース内の97の固有分類群(9属(種まで同定されていない配列) + 88種) を対象にin silicoテストを行った場合、85%(82/97)について増幅が確認できました。また、そのうち78% (64/82)が区別して同定可能であることが分かりました(Fig. 3)。

(灰)の分類群はin silico PCRで増幅されなかったもの。(濃い青色)は増幅が確認され、識別可能であったもの、配列を共有していることから、(薄い青)は属レベルで同定可能で(オレンジ)は化レベルで同定可能であることを示しています。
75個体の組織片から抽出されたDNAをもとにCeph18Sで増幅確認のテストとDNAの配列決定をしました。Fig.4の32分類群のうち14分類群はGenBankに登録されていませんでした。

太字の学名は、GenBank に同じ分類学的解像度 (属または種) への参照配列があったことを示しています。(灰)未増幅。学名の色は、形態学的IDとBLAST IDの一致が、種レベル (濃い青)、属レベル (水色)、科レベル (オレンジ)、または他の分類レベル (赤) と一致したことを表し、~ (チルダ) は、この領域で分類群を明確に識別できなかったことを示します。
16s rRNAプライマーセットとの比較
Ceph18S, CephMLS, S_CephalopodaのBc(データベース内の配列に対して増幅できた分類群の割合)とBs(データベース内の配列に対して種を同定できた割合)は下記の通りで、Ceph18Sと比較すると、増幅できる分類群の割合は同程度であるが、種を同定可能な割合が11-34%程低いという結果でした。
要は論文作成時の状態でそれぞれのプライマーを独立で使用したときに、CephMLS, S_Cephalopodaは種を識別する分解能がCeph18Sより低いということです。
| Primer | 領域 | DB | Bc | Bs | 登録されている分類群数 |
| Ceph18S | 18S rRNA | GenBank | 0.8 | 0.8 | 107 |
| 18S rRNA | SILVA | 0.85 | 0.78 | 97 | |
| CephMLS | 16S rRNA | – | 0.82 | 0.69 | 367 |
| S_Cephalopoda | 16S rRNA | – | 0.72 | 0.46 | 367 |
もう少し、視覚的にわかりやすくベン図で表すと以下のようになります。
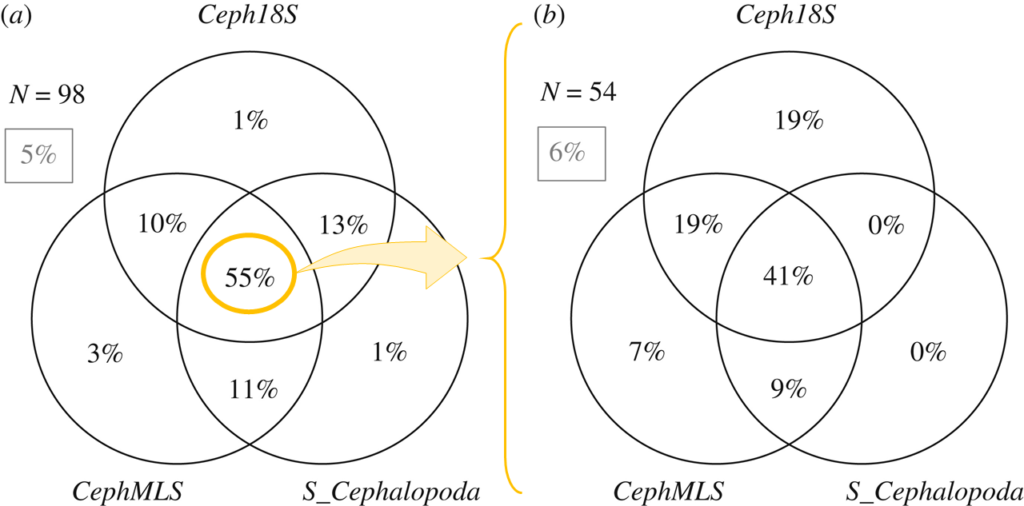
Fig.5は3つのプライマーを用いた時、データベースに登録されている頭足類の95%を増幅可能(a)で、94%が同定可能(b)でした。また、(a)16Sと18SのGenBank参照データベースの両方に存在した分類群の数(N)に対して、増幅可能だった分類群の割合を表しています。(b)3つのプライマーセットすべてで増幅された分類群(N)に対して、同定可能であった分類群の割合を表しています。
また、枠内の灰色の割合は、いずれのプライマーセットでも(a)増幅および(b)同定ができなかった分類群の数です。
Discussion
各章の抜粋です。
野外調査におけるCeph18Sの適用
新規開発されたCeph18Sは、核DNAの18S rRNA V2可変領域に隣接している比較的保存されている領域に設計しました。
増幅産物長が現在把握されている中で、131-196bpと可変的であることから、OTUなどにクラスタリングせずに、DADA2などでASVを生成してそのままデータベースと照合することでより高い解像度で結果を得ることができます。
Ceph18Sは全頭足類の44-66%(Bc×Bs)を増幅して同定できると推定されました。これに対して、S_CephalopodaとCephMLSは33%と56%と推定されています。
ただ、Ceph18Sはタコ類(Sepiids, Myopsids, Octopotheuthids, Gonatids)の検出には適していないことがin silicoによるテストと実験による検証で示唆されているので注意してください。
参照シーケンスについて
種類が知られている頭足類は約700種いる中で、18s rRNAの配列がSILVAには88種のみ、GenBankには完全長または部分長で300種程度しか存在していません。
多くの種を網羅していて高品質な参照データベースは、偽陰性(生息しているのに検出されない)や誤認(間違った同定)を減少させることができます。
そのためには、追加で種を同定し、配列を決定することが重要です。
複数プライマーによる生物多様性調査
既知の頭足類700種に対して、Ceph18SのBc(増幅できた割合)×Bs(種を同定できた割合)の値を当てはめてみると、Ceph18Sは約310-460種が増幅・同定できると推定される。
また、3つのプライマーすべてで増幅された分類群のうち、19%はCeph18Sでのみ、16%は16s rRNAのプライマーでのみ同定可能でした。
したがって、複数のユニバーサルプライマーの使用は検出できる分類群のカバー率と同定精度を高めることができます。
まとめ
まとめると
- 増幅効率が従来のプライマーと同程度以上で解像度が高いCeph18Sを開発した
- 増幅産物長が200bp程度と比較的短く、環境DNAには最適な長さ
- 3つのプライマーを使用すると増幅可能な分類群と同定可能な分類群が向上する
- GenBankに存在しない13種の頭足類の18s rRNAの配列を追加した
ということみたいです。
画像はジャーナルのCC BYに従う形で、記事で紹介したオープンアクセスの論文中の画像または、自身で作成したものを使用しています。





