
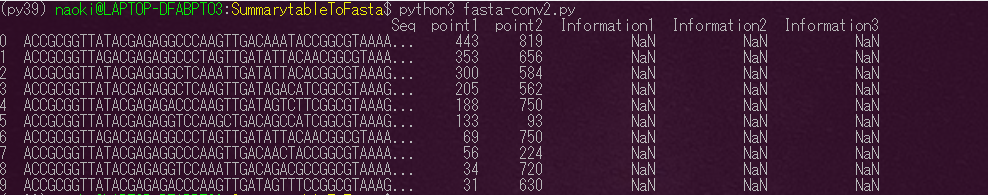
データ処理してると下記のような各地点で検出された配列とそれに対応するリード数が記載された状態のファイルを目にすることがあると思います。よくSummary tableといわれたりします。
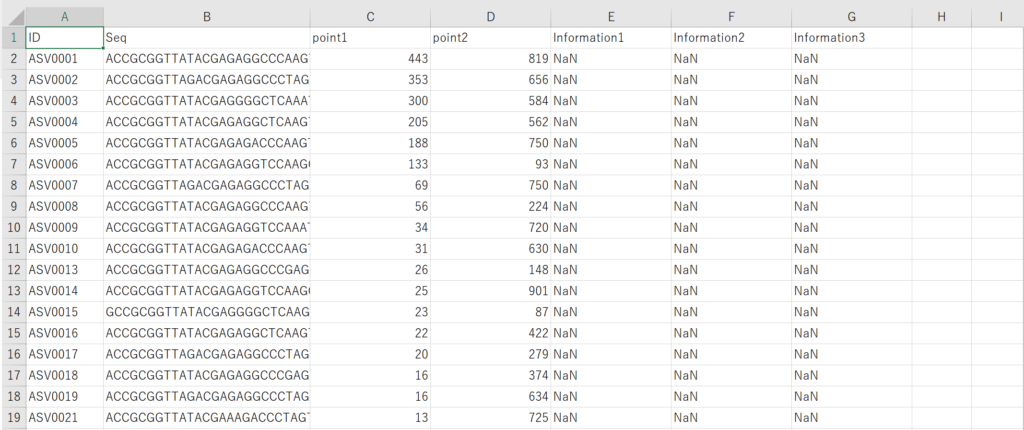
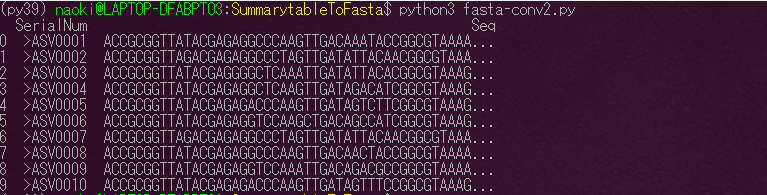
配列情報は以下のようなFasta形式にしておくことで相同性検索や、系統樹の作成などほかの解析に使用することができるようになります。

このようなデータの形式の変換は手でやってもいいですが、処理は可能な限り自動でやるほうが効率がいいので、今回はpythonを使ってSummary tableからIDと配列情報をセットで抽出してFastaファイルを作ってみたいと思います。
動作環境
- WSL
- Ubuntu 20.04 LTS
- Python 3.9.1
- Pandas
Python環境の構築に関しては下記記事を参照していただけると環境構築はできると思います。

表記ルール
コマンドラインにコードを書き込んでpythonスクリプトを実行する場合、下記のように先頭に$を表記します。なので$以降がコマンドです。
|
1 |
$ python3 fasta-conv.py |
pythonスクリプトファイルに書き込みを行う場合、下記のように$無しで表記します。
# が先頭にある行はコメントです
|
1 2 |
# パッケージのインポート import pandas as pd |
仮想環境
私はブログを書くときはpy39という仮想環境内で作業をしているのでpy39に入っておきます。
起動した状態は以下のような感じでbaseとなっています。

activateして仮想環境に入ります。
|
1 |
$ conda activate py39 ←ここは好きな仮想環境名 |
そうすると、py39という名前の仮想環境に入ることができます。
Summary tableの配列情報を抽出してFasta形式に変換する
冒頭に示したような形式のcsvファイルがを用意します。
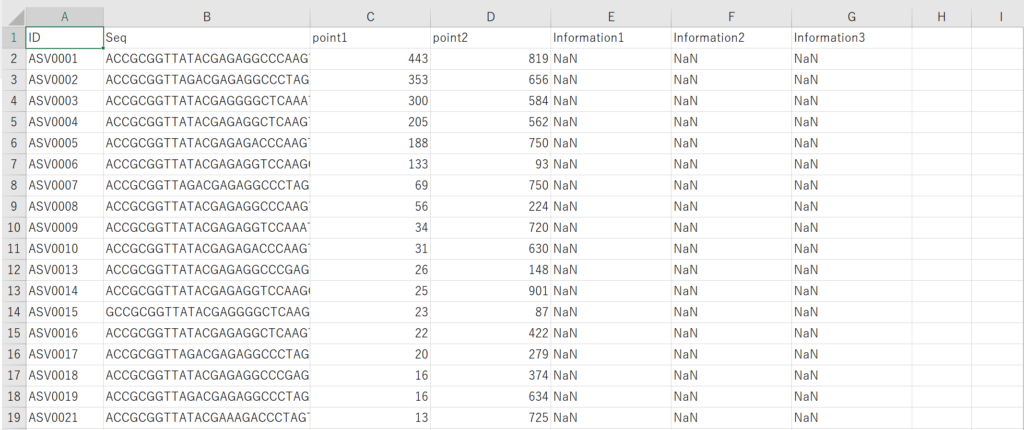
もし手元にない場合は以下のcsvファイルをダウンロードしてください。
pythonファイルの作成
用意したcsvファイルと同じフォルダにfasta-conv.pyを作成します。
作成できたら、control + LでパスWindowへ移り、bashと入力してEnterをクリックするとコマンドウィンドウが開けると思います。
あとはfasta-conv.pyをVSCodeなどのエディターで開いたら準備完了です。
パッケージのインポート
先ほど作成したfasta-conv.pyに下記コードを入力します。
|
1 |
import pandas as pd |
もしpandasをインストールしていない場合、
|
1 |
$ conda insatll pandas |
か
|
1 |
$ mamba install pandas |
でインストールしておきましょう。
データの読み込みと整形
まずはPandasを使ってデータフレーム形式でsummary tableファイルを読み込みます。扱うファイルはcsvファイルを前提として進めます。下記コードをfasta-conv.pyに入力しましょう。
|
1 2 |
# データフレームのインポート df = pd.read_csv('Summary_table.csv', header = 0, encoding = 'UTF-8') |
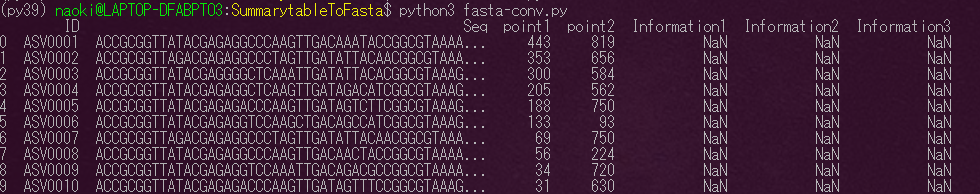
print関数をスクリプトに記入して確認してみましょう。
|
1 |
print(df.head(10)) |
コマンドラインで以下を実行
|
1 |
$ python3 fasta-conv.py |
ファイルの読み込みができたのでIDに>を加えた新しい行を追加します。
|
1 2 3 |
# >を各ASVのIDに追記 df['SerialNum'] = '>' + df['ID'].astype(str) print(df['SerialNum']) |
確認するならコマンドラインで以下を実行
|
1 |
$ python3 fasta-conv.py |

>を追記した列と配列が記載されている2列を列名を指定して抽出します
|
1 2 3 |
# SerialNumとSeqの列のみを抽出 df = df.loc[:,['SerialNum','Seq']] print(df.head(10)) |
確認する場合コマンドラインで以下を実行
|
1 |
$ python3 fasta-conv.py |
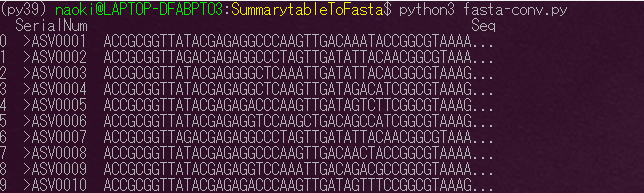
これで配列とセットとなるIDが抽出できました。
Fasta形式への変換と出力
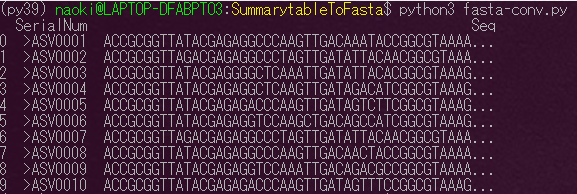
この縦に長い状態のデータをFasta形式で出力します。
Fasta形式は下記のような形式のファイルです。
>ASV0001 ←(配列の情報やID)
ACCTGCAATGCATGCAGTCGATCGATCGAT ←(塩基配列)
pandasのデータフレームはpandasのto_csv関数で出力します。関数名にcsvとついていますが、区切り文字を指定することで、カンマ区切り以外でも出力できます。
|
1 2 |
#改行を区切り文字として指定して拡張子を.fastaで出力 df.to_csv('ASVsequence.fasta', sep = '\n', index = None, header = None) |
今まで.pyファイル内に記載してきたprint()の部分は必要ないので#を先頭につけてコメントアウトしておきましょう。
fasta-conv.pyを実行すると、ASVsequence.fastaが同じフォルダに出力されます。
|
1 |
$ python3 fasta-conv.py |
fasta-conv.py まとめコード
|
1 2 3 4 5 6 7 8 9 10 11 12 |
import pandas as pd # データフレームのインポート df = pd.read_csv('Summary_table.csv', header = 0, encoding = 'UTF-8') #print(df.head(10)) # >を各ASVのIDに追記 df['SerialNum'] = '>' + df['ID'].astype(str) # print(df['SerialNum']) # SerialNumとSeqの列のみを抽出 df = df.loc[:,['SerialNum','Seq']] #print(df.head(10)) #改行を区切り文字として指定して拡張子を.fastaで出力 df.to_csv('ASVsequence.fasta', sep = '\n', index = None, header = None) |
番外編1 : 配列に任意のIDを付与してからFasta形式で出力する
下記のように配列情報だけでIDが降られてないSummary tableデータが手元にある場合を考えてみます。
それぞれの配列に任意のIDを振ってFastaファイルを出力してみましょう。
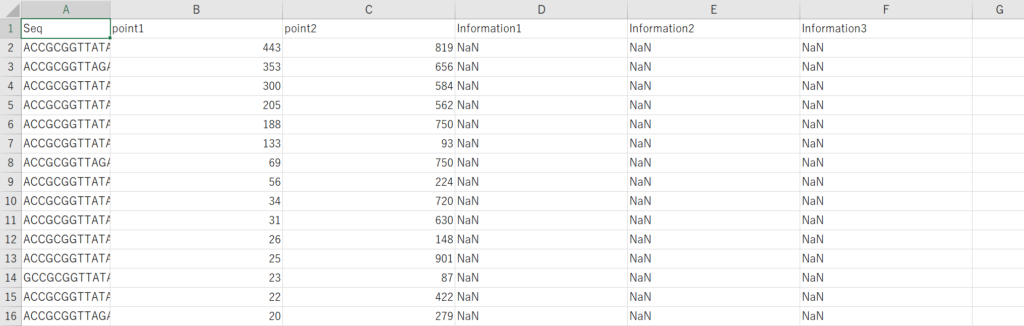
冒頭で用意したSummary tableのIDの列だけを除去したcsvファイル(Summary_table2.csv)を用意します。
Pythonファイルの作成
fasta-conv2.pyを作成します。
次に、control + LでパスWindowへ移り、bashと入力してEnterをクリックするとコマンドwindowが開けると思います。
fasta-conv2.pyをVSCodeなどの好きなエディターで開いたら準備完了です。
パッケージのインポート
先ほど作成したfasta-conv2.pyに下記コードを入力します。
パッケージのインポート
|
1 |
import pandas as pd |
データの読み込みと整形
Pandasのread_csv関数でSummary_table2.csvを読み込みます。
|
1 2 3 |
# データフレームのインポート df = pd.read_csv('Summary_table2.csv', header = 0, encoding = 'UTF-8') print(df.head(10)) |
確認する場合コマンドラインで以下を実行
|
1 |
$ python3 fasta-conv2.py |
次に配列の数だけ通し番号を振ります。
|
1 2 |
# 開始を1, 終了を行数として1ずつ増加させる df['SN'] = pd.RangeIndex(start = 1, stop = len(df) + 1, step = 1) |
各配列に対して管理しやすいように、通し番号を利用したIDを付与します。
zfill関数は0埋めをしてくれる関数で、下記のように4を指定している場合、1であれば0001、10であれば0010という風になります。
|
1 2 3 |
# >ASVと0埋めした通し番号を合わせた新しい列を作成 df['SerialNum'] = '>ASV' + df['SN'].astype(str).str.zfill(4) print(df['SerialNum']) |
確認する場合コマンドラインで以下を実行
|
1 |
$ python3 fasta-conv2.py |

新しく作成したSerialNumと配列が記載されているSeq列を抽出します。
|
1 2 3 |
# SerialNumとSeq列を抽出 df = df.loc[:,['SerialNum','Seq']] print(df.head(10)) |
確認する場合コマンドラインで以下を実行
|
1 |
$ python3 fasta-conv2.py |
最後にpandasのto_csv関数で、fasta形式のファイルを出力します。
|
1 2 |
#改行を区切り文字として指定して拡張子を.fastaで出力 df.to_csv('ASVsequence2.fasta', sep = '\n', index = None, header = None) |
fasta-conv2.py まとめコード
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 |
import pandas as pd # データフレームのインポート df = pd.read_csv('Summary_table2.csv', header = 0, encoding = 'UTF-8') #print(df.head(10)) # 開始を1, 終了を行数として1ずつ増加させる df['SN'] = pd.RangeIndex(start = 1, stop = len(df) + 1, step = 1) # >ASVと0埋めした通し番号を合わせた新しい列を作成 df['SerialNum'] = '>ASV' + df['SN'].astype(str).str.zfill(4) #print(df['SerialNum']) # SerialNumとSeq列を抽出 df = df.loc[:,['SerialNum','Seq']] print(df.head(10)) #改行を区切り文字として指定して拡張子を.fastaで出力 df.to_csv('ASVsequence2.fasta', sep = '\n', index = None, header = None) |
番外編2 : Web BLASTで相同性検索してみる
Summary tableの形式にまとめられた各ASVやOTU配列がなんの生物種に由来するのか気になると思うので検索してみましょう。
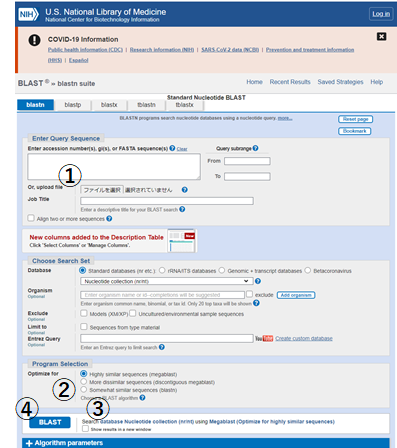
NCBIのNucleotide BLASTのページで操作します。
先ほど作成したFastaファイルを選択①して、blastnを選択②したのち、別windowに出力するオプションにチェック③を入れたらBLAST検索④を開始します。
私の環境では1分くらいで結果が出力されました。
Blast検索の結果の見方
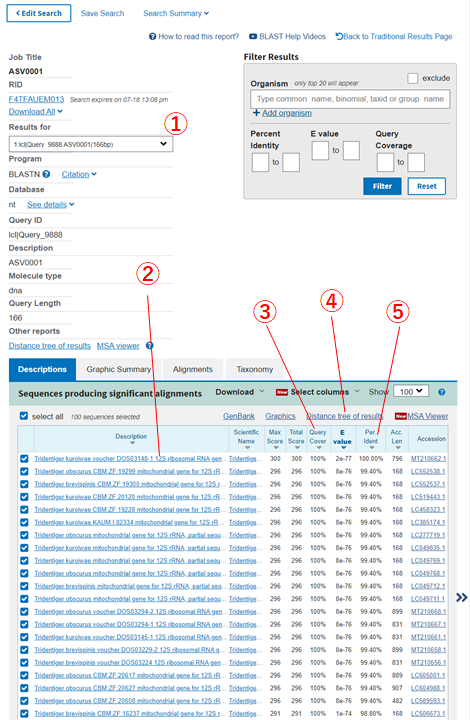
いつも私がみている情報は以下の5点です。
- 選択した.fastaファイル内の配列情報がここに表示されます
- ①で選択した配列にHitしたデータベースの登録種名情報です
- ①で選択した配列とデータベースの配列の一致割合です
100%の配列を優先して結果を見ます - ①で選択した配列とデータベースの配列を用いて、系統樹を作成できます
同じクレード内に別種が含まれていないか、配列を共有している種がいないかを確認します - ①で選択した配列がデータベースの配列とどの程度一致しているか(何%の相同性か)が表示されます
上記情報より各配列がどの種に由来するものなのかを推測していきます。
まとめ
- pythonを使ってsummry table形式のデータから配列情報を抜き出してfasta形式で得る方法について解説しました。
- 作成したpythonスクリプトで得られたFastaファイルを使ってWebのBLAST検索を行いました。
免責事項
十分注意は払っていますが、本記事の情報・内容について保証されるものではありません。また、本記事の利用や閲覧によって生じたいかなる損害について責任を負いません。そして、本記事の情報は予告なく変更される場合がありますので、ご理解くださいますようお願い申し上げます。





