
QIIME2はアンプリコンシーケンスのデータ解析を中心に現在もサポートが続いているデータ解析パイプラインです。またQIIME2と聞くと菌叢解析を想像しますが専用品ではないのでMiseqなどのショートリードシーケンサーから得られたデータであれば好きな解析が可能です。
今回は手持ちのデータがなくても解析できるように、先行研究で配列決定された魚類の環境DNAシーケンスデータをSRA toolkitで取得後、QIIME2でデータ解析をしたのちにBlast+で種のアサイメントを実施します。
SRA toolkitを用いた配列取得とQIIME2での解析、Blast+での種のアサイメントは独立なので、例えば配列取得がうまく行かなくてもfastq.gzさえ手元にあればQIIME2以降の解析は可能です。
動作環境
- Ubuntu 22.04 LTS
(多分WSL, WSL2 + Ubuntuでも同様に動作すると思います) - python 3.8
- SRA toolkit : github
- fasterq-dump version 3.0.2
- prefetch version 3.0.2
- BLAST+ version 2.13: github
SRA toolkitは以前の記事からversionが上がっている点に注意してください。最新のものに置き換えて使用されるPCにインストールください。

またDesktop上にqiime2というフォルダを作成して、そこを中心にファイル生成を行います。
デスクトップ上で、ターミナルを開いてqiime2フォルダや、その他の必要フォルダの作成をしておきましょう。
|
1 2 3 4 5 6 |
# フォルダの作成 mkdir -p ./qiime2/01output/qza ./qiime2/01output/qzv ./qiime2/01output/other ./qiime2/02ref ./qiime2/03log # 作成したディレクトリに移動 cd qiime2 # このような感じなっていたらOK→ (base) (username)@(PC name):~/Desktop/qiime2 |
Miniconda3を使ってQiime2をインストール
パッケージマネージャーとしてcondaを提供するMiniconda3を使ってQIIME2をインストールします(QIIME2の推奨法)。Minicondaのインストールは以下の記事を参考に自分の環境に合ったものを選択してください。

QIIME2のインストールはQIIME2のチュートリアル(https://docs.qiime2.org/2022.11/install/native/)に沿って進めます。2022年12月末現在のQIIME2のバージョンは2022.11です。
Minicondaのアップデート
とりあえずcondaのアップデートを行います。ターミナルを開いて以下のコマンドでアップデート。
|
1 |
conda update conda |
# All requested packages already installed.と表示されていたらアップデートは終わってます。
mambaのインストール
チュートリアルにはありませんが、パッケージマネージャーとして優秀なmambaをインストールしておきます。
|
1 |
conda install -c bioconda mamba -y |
wgetのインストール
ダウンローダーとしてwgetを使用するので、mambaでwgetをインストールしておきます。
|
1 |
mamba install wget -y |
qiime2 core 2022.11 distributionをcondaの仮想環境にインストール
以前にQiime2をインストールしたことがある、もしくはインストールを試みたことがある場合も含め、各リリース専用の仮想環境を構築して運用することを推奨します。
以前のバージョンは2022.8でしたが2022.11になっていくつか機能追加がされているようです(次のリリースは2023.2)。
では、仮想環境としてqiime2-2022.11を作成していきましょう。まず、wgetでパッケージバージョンなどが記載されたymlファイルをダウンロードします。
|
1 |
wget https://data.qiime2.org/distro/core/qiime2-2022.11-py38-linux-conda.yml |
lessなどで中身を見ると、qiime2-2022.11-py38-linux-conda.ymlにはQIIME2での解析に必要な基本的なライブラリがバージョンとともに記載されています。
ymlファイルの情報を使って、2022.11バージョンの仮想環境を作成します。
|
1 |
mamba env create -n qiime2-2022.11 --file qiime2-2022.11-py38-linux-conda.yml |
特にエラーなく以下のような指示が出たらOK。
|
1 2 3 4 5 6 7 |
# To activate this environment, use # # $ conda activate qiime2-2022.11 # # To deactivate an active environment, use # # $ conda deactivate |
では、指示に従って仮想環境に入っておきましょう。
|
1 2 |
mamba activate qiime2-2022.11 # このような感じなっていたらOK→ (qiime2-2022.11) (username)@(PC name):~/Desktop/qiime2 |
仮想環境を出るときは、mamba deactivate、仮想環境がどんな名前だったか確認したい時はmamba info -eで確認できます。最後に使用したymlファイルはいらないので削除。
|
1 |
rm qiime2-2021.11-py38-linux-conda.yml |
ここれでQIIME2のインストールは完了。
また、ターミナル上で作業を進めるのであれば、最初に以下のコマンドを実行しておくとqiimeのコマンドをタブ補完してくれます(2023/4/8 追記)
|
1 |
source tab-qiime |
後で使用するライブラリをインストールしておく
|
1 |
mamba install seqkit parallel pigz -y |
次はSRA toolkitで先行研究で配列決定されたデータをダウンロードします。
SRA toolkitを使ってハイスループットシーケンスデータを取得する
今回は2022年に宮先生が市民科学×環境DNAというテーマで実施された、MiFishプライマーによる魚類群集構造解析論文のデータを使って進めていきたいと思います。
沿岸海域における地域の生物多様性を評価するための魚の eDNA メタバーコーディングにおける市民科学の使用: パイロット研究
The use of citizen science in fish eDNA metabarcoding for evaluating regional biodiversity in a coastal marine region: A pilot study
DOI : 10.3897/mbmg.6.80444
文中には以下のようにあるので、NCBIのSRAデータベースより登録されているデータのアクセッション番号(SRR〜)を確認します。
All raw DNA sequence data and associated information were deposited in the DDBJ/EMBL/GenBank database and are available under accession number DRA012840.
確認したところ、DRA012840には6つのデータがあってDRR321580~DRR321585が割り振られているのでその情報を使って配列データを取得します。
prefetch によるSRAデータのダウンロード
prefetchはSRAデータをダウンロードしてくるコマンドです。一度.sraファイルをダウンロードしておくとfastqへの変換に失敗してしまったりした際にやり直しが楽です。
DRR321580~DRR321585の配列データを一括でダウンロードします。
|
1 |
for i in `seq 80 85`; do prefetch -p DRR3215$i --output-file ./output/DRR3215$i.sra ;done |
実行するとoutputフォルダが作成され、その中にDRR~.sraという形式で6データがダウンロードされます。次はfastqerq-dumpを使ってダウンロードした.sraファイルからFastqファイルを生成します。
fasterq-dumpによるsraファイルからのfastq生成
先程outputフォルダに生成した.sraファイルをもとにfastqファイルを00fastqgzフォルダに生成します。
|
1 |
for i in `seq 80 85` ; do fasterq-dump -e 24 /output/DRR3215$i.sra --outdir ./00fastqgz/ ;done |
オプションを2つ使用しています。-pは進捗状況を表示、-eは複数のコアを使用しての並列処理オプションです。
_1.fastqと_2.fastq はイルミナのHTSから出力された状態だとR1, R2にあたるファイルです。FowardプライマーとReverseプライマーで読まれた配列になります。
- DRR32158X_1.fastq
- DRR32158X_2.fastq
.fastqファイルはpigzで圧縮した後に名前を_1 →_R1、_2 → _R2に変換しておきます。
|
1 2 3 4 5 |
# 圧縮 pigz ./00fastqgz/*.fastq # 変換 rename -E 's/_1/_R1/g' ./00fastqgz/*_1.fastq.gz rename -E 's/_2/_R2/g' ./00fastqgz/*_2.fastq.gz |
00fastqgzフォルダの中身がDRR32158X_R1.fastq.gzとDRR32158X_R2.fastq.gzとなっていればOKです。
QIIME2によるデータ処理
ここからは、SRA-toolkitでダウンロードした配列データを用いてQIIME2によるデータ解析処理を進めていきます。ざっくりいうと以下のような感じです。
- FastqファイルをQIIME2へインポート
- seqfu
- qiime tools import
- Demultiplex (シーケンスをサンプルに配分)
- 配列上の非生物学的部分(ex. primer)の除去
- qiime cutadapt trim-paired
- Quality scoreの可視化
- qiime demux summarize
- クオリティフィルタリング、デノイジング、キメラ除去
- qiime dada2 denoise-paired
- qiime feature-table filter-samples
- qiime feature-table filter-seqs
- データのエクスポート
- qiime tools export
- biom convert
Manifestファイルの作成とQiime2へのデータのインポート
QIIME2へfastqファイルをインポートする際、ファイル情報をまとめたmanifestファイルが必要になります。
MGIやPacBioなどのシーケンサーから出力されたfastqファイルはどうか分かりませんが、最近Illunaのシーケンサーから取得したようなfastqファイルであれば、–typeにはSampleData[PairedEndSequencesWithQuality]を–input-formatにはPairedEndFastqManifestPhred33V2を指定すれば良いかと思います。
| Read type | Phred score | –type | –input-format | 備考 |
|---|---|---|---|---|
| Single-end | PHRED 33 | SampleData[SequencesWithQuality] | SingleEndFastqManifestPhred33V2 | |
| Paired-end | PHRED 33 | SampleData[PairedEndSequencesWithQuality] | PairedEndFastqManifestPhred33V2 | |
| Single-end | PHRED 64 | SampleData[SequencesWithQuality] | SingleEndFastqManifestPhred64V2 | |
| Paired-end | PHRED 64 | SampleData[PairedEndSequencesWithQuality] | PairedEndFastqManifestPhred64V2 | |
| Paired-end | PHRED 64 | SampleData[PairedEndSequencesWithQuality] | CasavaOneEightSingleLanePerSampleDirFmt | fastqファイルの形式が.+_.+_L[0-9][0-9][0-9]_R[12]_001\.fastq\.gzである必要あり |
指定できる値は以下のコマンドで確認できます。
|
1 2 3 4 |
# --typeに指定できる値を確認 qiime tools import --show-import-types # --input-formatに指定できる値を確認 qiime tools import --show-import-formats |
Manifestファイルはawkコマンドを使用して作成することもできますがseqfuを使うと簡単です。両者はManifestファイルの形式が少し異なる点に注意です。
開発者githubはこちら。オプションは以下の通り。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 |
SeqFu 1.17.1 - FASTX Tools ____ _____ / ___| ___ __ _| ___| _ \___ \ / _ \/ _` | |_ | | | | ___) | __/ (_| | _|| |_| | |____/ \___|\__, |_| \__,_| |_| · bases : count bases in FASTA/FASTQ files · check : check FASTQ file for errors · count [cnt] : count FASTA/FASTQ reads, pair-end aware · deinterleave [dei] : deinterleave FASTQ · derep [der] : feature-rich dereplication of FASTA/FASTQ files · interleave [ilv] : interleave FASTQ pair ends · lanes [mrl] : merge Illumina lanes · list [lst] : print sequences from a list of names · metadata [met] : print a table of FASTQ reads (mapping files) · rotate [rot] : rotate a sequence with a new start position · sort [srt] : sort sequences by size (uniques) · stats [st] : statistics on sequence lengths · cat : concatenate FASTA/FASTQ files · grep : select sequences with patterns · head : print first sequences · rc : reverse complement strings or files · tab : tabulate reads to TSV (and viceversa) · tail : view last sequences · view : view sequences with colored quality and oligo matches Type 'seqfu version' or 'seqfu cite' to print the version and paper, respectively. Add --help after each command to print its usage. Unknown subprogram: -h |
|
1 |
seqfu metadata --format manifest --threads 24 00fastqgz > Manifest.tsv |
Manifest.tsvは以下のようにな形式になっているはずです。
| sample-id | forward-absolute-filepath | reverse-absolute-filepath |
|---|---|---|
| DRR321580 | $HOME/Desktop/qiime2/00fastqgz/DRR321580_R1.fastq.gz | $HOME/Desktop/qiime2/00fastqgz/DRR321580_R2.fastq.gz |
| DRR321581 | $HOME/Desktop/qiime2/00fastqgz/DRR321581_R1.fastq.gz | $HOME/Desktop/qiime2/00fastqgz/DRR321581_R2.fastq.gz |
| DRR321582 | $HOME/Desktop/qiime2/00fastqgz/DRR321582_R1.fastq.gz | $HOME/Desktop/qiime2/00fastqgz/DRR321582_R2.fastq.gz |
| DRR321583 | $HOME/Desktop/qiime2/00fastqgz/DRR321583_R1.fastq.gz | $HOME/Desktop/qiime2/00fastqgz/DRR321583_R2.fastq.gz |
| DRR321584 | $HOME/Desktop/qiime2/00fastqgz/DRR321584_R1.fastq.gz | $HOME/Desktop/qiime2/00fastqgz/DRR321584_R2.fastq.gz |
| DRR321585 | $HOME/Desktop/qiime2/00fastqgz/DRR321585_R1.fastq.gz | $HOME/Desktop/qiime2/00fastqgz/DRR321585_R2.fastq.gz |
ちなみに、先程のURL先のページを参考にawkコマンドでManifest.csvが作成できます。
|
1 2 3 |
ls ./00fastqgz/* | \ awk -F '[/_]' -v P="$(pwd)" 'BEGIN {print "sample-id,absolute-filepath,direction"} \ {if(NR%2==0){print $3","P"/"$3_$4",reverse"}else{print $3","P"/"$3_$4",forward"}}' > Manifest.csv |
上記コマンドで作成したManifest.csvは以下のようにな形式になっているはずです。
| sample-id | absolute-filepath | direction |
|---|---|---|
| DRR321580 | $HOME/Desktop/qiime2/00fastqgz/DRR321580_R1.fastq.gz | forward |
| DRR321580 | $HOME/Desktop/qiime2/00fastqgz/DRR321580_R2.fastq.gz | reverse |
| DRR321581 | $HOME/Desktop/qiime2/00fastqgz/DRR321581_R1.fastq.gz | forward |
| DRR321581 | $HOME/Desktop/qiime2/00fastqgz/DRR321581_R2.fastq.gz | reverse |
| DRR321582 | $HOME/Desktop/qiime2/00fastqgz/DRR321582_R1.fastq.gz | forward |
| DRR321582 | $HOME/Desktop/qiime2/00fastqgz/DRR321582_R2.fastq.gz | reverse |
| DRR321583 | $HOME/Desktop/qiime2/00fastqgz/DRR321583_R1.fastq.gz | forward |
| DRR321583 | $HOME/Desktop/qiime2/00fastqgz/DRR321583_R2.fastq.gz | reverse |
| DRR321584 | $HOME/Desktop/qiime2/00fastqgz/DRR321584_R1.fastq.gz | forward |
| DRR321584 | $HOME/Desktop/qiime2/00fastqgz/DRR321584_R2.fastq.gz | reverse |
| DRR321585 | $HOME/Desktop/qiime2/00fastqgz/DRR321585_R1.fastq.gz | forward |
| DRR321585 | $HOME/Desktop/qiime2/00fastqgz/DRR321585_R2.fastq.gz | reverse |
seqfu, awkともに今回のデータはPaired-end sequenceデータなのでforwardとreverseの絶対パスが表記されています。
ManifestファイルができたらQIIME2に配列データをインポートします。seqfuで作成したtsv形式のManifestファイルを使用する場合、–input-formatの語尾にV2を、awkコマンドベースで作成したようなcsv形式のManifestファイルの場合は–input-formatの語尾にV2をつけなくて大丈夫です。
Plug in : qiime tools import
|
1 2 3 4 5 |
qiime tools import \ --type 'SampleData[PairedEndSequencesWithQuality]' \ --input-path Manifest.tsv \ --input-format PairedEndFastqManifestPhred33V2 \ --output-path 01output/qza/00demux.qza |
|
1 |
Imported Manifest.tsv as PairedEndFastqManifestPhred33V2 to 01output/qza/00demux.qza |
と出ていればOKです。
Cutadaptによるプライマー除去とデータの視覚化
以降は、一般的にDADA2によるデノイジングに移ることが多いです。
DADA2は5’末端からプライマー部分の配列が固定長である場合、5’末端から指定した配列長を除去する–p-trun-left-f, –p-trun-left-rオプションでプライマー配列の除去が可能です。ただ、今回は複数プライマー(MiFish-U, U2, Ev2)を使用しているということもあり、cutadaptによってプライマー配列を除去するステップを追加します。
この論文で使用しているプライマーは以下の通り
| プライマー名 | 配列(5’→3′) | 配列長 |
| MiFish-U-F (硬骨魚類) | NNNNNNGTCGGTAAAACTCGTGCCAGC | 27 |
| MiFish-U-R (硬骨魚類) | NNNNNNCATAGTGGGGTATCTAATCCCAGTTTG | 33 |
| MiFish-Ev2-F (軟骨魚類) | NNNNNNRGTTGGTAAATCTCGTGCCAGC | 28 |
| MiFish-Ev2-R (軟骨魚類) | NNNNNNGCATAGTGGGGTATCTAATCCTAGTTTG | 34 |
| MiFish-U2-F (アナハゼ類) | NNNNNNGCCGGTAAAACTCGTGCCAGC | 27 |
| MiFish-U2-R (アナハゼ類) | NNNNNNCATAGGAGGGTGTCTAATCCCCGTTTG | 33 |
| プライマー名 | 配列(5’→3′) | 配列長 |
| MiFish-Unity-F (コンセンサス配列) | NNNNNNGYYGGTAAAWCTCGTGCCAGC | 27 |
| MiFish-Unity-R (コンセンサス配列) | NNNNNNCATAGKRGGGTRTCTAATCCYMGTTTG | 33 |
今回は複数プライマーの配列をもとに変異のある部分は縮合塩基に置き換えたコンセンサス配列を指定します。(実際にプライマー配列を指定する際、NNNNNNを入れなかたのは生データをseqfu viewで確認した際、N部分の塩基数がバラバラであったためです:備考の章を参照)
Plug in : qiime cutadapt trim-paired
|
1 2 3 4 5 6 7 8 |
qiime cutadapt trim-paired \ --i-demultiplexed-sequences ./01output/qza/00demux.qza \ --p-front-f GYYGGTAAAWCTCGTGCCAGC \ --p-front-r CATAGKRGGGTRTCTAATCCYMGTTTG \ --o-trimmed-sequences ./01output/qza/01primer-trimmed-demux.qza \ --p-discard-untrimmed true \ --p-cores 24 \ --verbose |
|
1 |
Saved SampleData[PairedEndSequencesWithQuality] to: 01output/qza/01primer-trimmed-demux.qza |
と出ていればOKです。
使用しているオプションのうち–p-discard-untrimmedは、プライマー配列を認識できなかったリードは廃棄するオプションです。ライブラリ調整や配列決定時のエラーが発生していそうなものは廃棄したいので使用しました。
Quality scoreの可視化
QIIME2は分析に使用するqzaファイルの他に、qiime demux summarizeでqzaファイルを変換した可視化用ファイルqzvが利用できます。生成されたqzvファイルはqiime2 viewで確認できます。
importした配列データ(00demux.qza)とCutadaptで処理した後(01primer-trimmed-demux.qza)のクオリティプロットをqzvファイルを生成して比較してみます。
Plug in : qiime demux summarize
importしたデータ
|
1 2 3 |
qiime demux summarize \ --i-data ./01output/qza/00demux.qza \ --o-visualization ./01output/qzv/00demux.qzv |
Cutadaptによるプライマー除去後の配列データ
|
1 2 3 |
qiime demux summarize \ --i-data ./01output/qza/01primer-trimmed-demux.qza \ --o-visualization ./01output/qzv/01primer-trimmed-demux.qzv |
|
1 2 |
Saved Visualization to: 01output/qzv/00demux.qzv Saved Visualization to: 01output/qzv/01primer-trimmed-demux.qzv |
qiime2 viewを開いて、
Drag and drop or click hereの部分に00demux.qzv, 01primer-trimmed-demux.qzvを入れてみると、Bar plotが表示されます。上段のInteractive Quality PlotタブをクリックするとR1, R2のQuality Plotが出てきます。
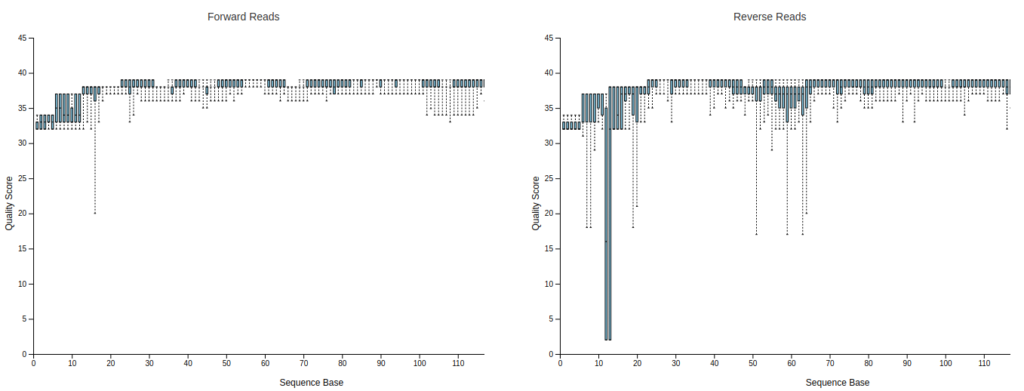
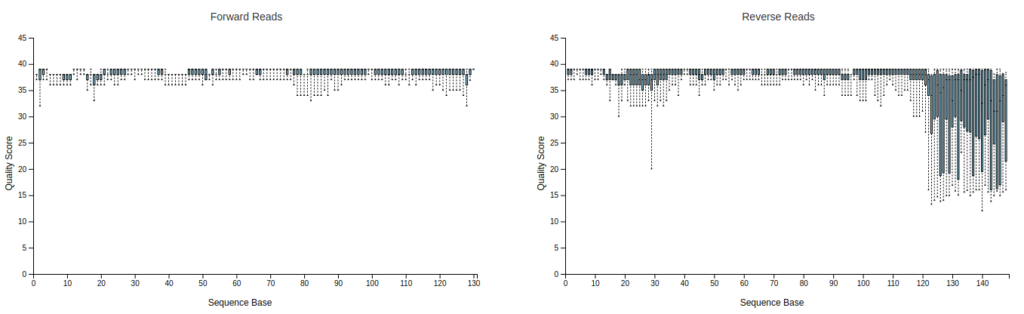
プライマー部位が除去されて151bpより短くなっているのが分かります。
DADA2によるデノイジング、エラー配列の修正
DADA2はクオリティスコアをエラーモデルに使用しているので、低品質な配列を切り取っておくとRare variantな配列の感度が向上するようです。
そのため、R1, R2ともに後半の配列(3’側)を–p-trunc-len-f, –p-trunc-len-rオプションで切り捨てます。また、Paired-endシーケンスは、Forwardプライマー側(R1)とReverseプライマー側(R2)つなげる必要があります。
最終的に、以下の図でいうところのRead1, Read2がInsertとかぶる部分の配列を残して結合したいので、ForwardとReverseプライマー配列が除去される(ている)ことと、種判別領域は可変長であることが多いことを念頭に置き、平均産物長をベースに–p-trunc-len-f, –p-trunc-len-rの値を決定します。

まとめると考慮すべき事項は以下の通り。
- Forward primer length 最大 27bp (NNNNNNを含む)
- Reverse primer length 最大 33bp (NNNNNNを含む)
- Overlap length >= 20bp (最低12bpまで可)
- R1よりR2のほうがクオリティが早く落ちやすいので、–p-trunc-len-f >= –p-trunc-len-rとしておく
- MiFishの種判別領域の平均産物長は172bp、軟骨魚類やヤツメ類などは180bp程度ある(経験則)
となると最低でも約180bp程度は残るほうが良さそうです。
R1はFプライマーを除くと最低で124bp、R2はRプライマーを除くと最低でも118bpは残る予定です。20bpのOverlap regionを加味しても222bpは残るので少し削って結合しても種判別領域は生成できそうです。
| 項目 | 配列長 | 残り (おおよそ) |
| サイクル数 | 151 + 151 = 302bp | 302bp |
| Fプライマー | 27bp | 275bp |
| Rプライマー | 33bp | 242bp |
| Overlap | 20bp | 222bp |
–p-trunc-len-f >= –p-trunc-len-rというルールは守りつつ222bp以上を目指すと、–p-trunc-len-fは115bp, –p-trunc-len-rも115bpとしてDADA2の解析を進めても問題なさそうです。
その他にも色々とオプションはありますが、指定の値以外はデフォルトで実施。
Plug in : qiime dada2 denoise-paired
|
1 2 3 4 5 6 7 8 9 10 11 |
qiime dada2 denoise-paired \ --i-demultiplexed-seqs 01output/qza/01primer-trimmed-demux.qza \ --p-trim-left-f 0 \ --p-trim-left-r 0 \ --p-trunc-len-f 115 \ --p-trunc-len-r 115 \ --o-representative-sequences ./01output/qza/02rep-seqs-dada2_f115_r115.qza \ --o-table ./01output/qza/02table-dada2_f115_r115.qza \ --o-denoising-stats ./01output/qza/02stats-dada2_f115_r115.qza \ --p-n-threads 0 \ --verbose |
|
1 2 3 |
Saved FeatureTable[Frequency] to: ./01output/qza/02table-dada2_f115_r115.qza Saved FeatureData[Sequence] to: ./01output/qza/02rep-seqs-dada2_f115_r115.qza Saved SampleData[DADA2Stats] to: ./01output/qza/02stats-dada2_f115_r115.qza |
リードフィルタリング
データを取得元のMiya et al.2020でもやっているので、Singleton, Doubleton, Tripletonを除くために4リード未満の配列を削除します。
Plug in : qiime feature-table filter-samples
|
1 2 3 4 |
qiime feature-table filter-features \ --i-table ./01output/qza/02table-dada2_f115_r115.qza \ --p-min-frequency 4 \ --o-filtered-table ./01output/qza/03frequency-filtered-table.qza |
|
1 |
Saved FeatureTable[Frequency] to: ./01output/qza/03frequency-filtered-table.qza |
リードフィルタリングしたテーブルデータをもとにフィルタリングされて生き残った配列データを出力します。
Plug in : qiime feature-table filter-seqs
|
1 2 3 4 |
qiime feature-table filter-seqs \ --i-data ./01output/qza/02rep-seqs-dada2_f115_r115.qza \ --i-table ./01output/qza/03frequency-filtered-table.qza \ --o-filtered-data ./01output/qza/03filtered-rep-seqs.qza |
|
1 |
Saved FeatureData[Sequence] to: ./01output/qza/03filtered-rep-seqs.qza |
データのエクスポート
配列データのエクスポート
配列データはdna-sequences.fastaとして出力指定したフォルダに出力されます。
Plug in : qiime tools export
|
1 2 3 |
qiime tools export \ --input-path ./01output/qza/03filtered-rep-seqs.qza \ --output-path ./01output/other/ |
|
1 |
Exported ./01output/qza/03filtered-rep-seqs.qza as DNASequencesDirectoryFormat to directory ./01output/other/ |
ファイル名の変更
|
1 |
mv ./01output/other/dna-sequences.fasta ./01output/other/Representative_seq.fasta |
Summary tableのエクスポート
Table形式のデータはBIOM形式のファイルとしてエクスポートされます。BIOM形式のデータはbiom convertによって.tsv形式のデータへ変換することが出来ます。
Plug in : qiime tools export, biom convert
リードフィルタリングする前のテーブルデータをエクスポート
|
1 2 3 |
qiime tools export \ --input-path ./01output/qza/02table-dada2_f115_r115.qza \ --output-path ./01output/other/ |
|
1 |
Exported ./01output/qza/02table-dada2_f115_r115.qza as BIOMV210DirFmt to directory ./01output/other/ |
biomファイルからtsvファイルへ
|
1 2 3 4 |
biom convert \ -i ./01output/other/feature-table.biom \ -o ./01output/other/02feature-table.tsv \ --to-tsv |
リードフィルタリングした後のテーブルファイルをエクスポート
|
1 2 3 |
qiime tools export \ --input-path ./01output/qza/03frequency-filtered-table.qza \ --output-path ./01output/other/ |
biomファイルからtsvファイルへ
|
1 2 3 4 |
biom convert \ -i ./01output/other/feature-table.biom \ -o ./01output/other/03feature-filtered-table.tsv \ --to-tsv |
|
1 |
Exported ./01output/qza/03frequency-filtered-table.qza as BIOMV210DirFmt to directory ./01output/other/ |
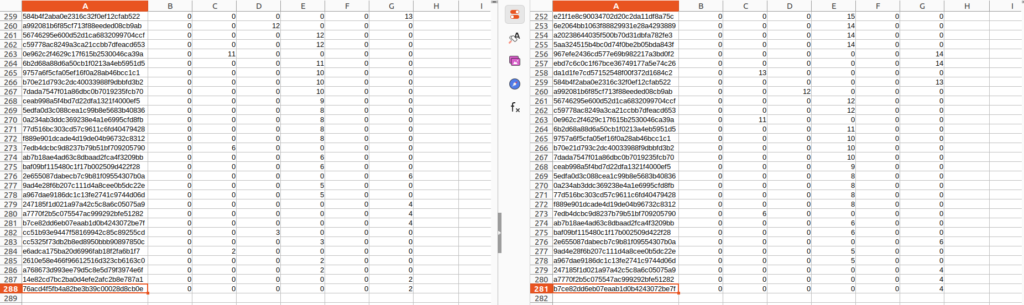
フィルタリングを行う前後で比較すると行数が減っていることが分かります。またこのテーブルデータを参考に出力した、Representative_seq.fastaの配列数もフィルタリング後のテーブルデータのASV数と同数になっているはずです。
ここまでこれば、得られた配列データに対して相同性検索をかけて種を推定することが出来ます。次はBlast+によるBLASTnを実施します。
BLASTnによる塩基配列の相同性検索
まず、魚類の12s rRNAの配列データを用意します。手元に配列データがない場合、2023/1/4にダウンロードしたものがこちらにあります(PMiFish用のデータベース形式になっています)。
データベース用のfastaファイルを02refフォルダに入れて、名前をMiFishDB.fastaに変更しておきます。コマンドで指定するのでそこを変えるなら他の名前でもOKです。
BLAST+のダウンロード
Blast+は配列の相同性検索を行うパッケージです。DNA配列に対してはBLASTnを実行します。mambaを使ってインストールします。
|
1 |
mamba install -c bioconda 'blast>=2.13' -y |
完了したら、確認。
|
1 |
blastn -h |
一番下に DESCRIPTION Nucleotide-Nucleotide BLAST 2.13.0+ と出ていたらよいです。インストールできない場合は、必要なcondaのchannelが登録されていないかもしれないので、確認・追加して再度実行してみてください。
|
1 2 3 |
conda config --add channels defaults conda config --add channels bioconda conda config --add channels conda-forge |
Local Blast (BLASTn) の実行
まずfasta形式のデータベースファイルからBLASTn用のデータベースファイル群を作成します。
makeblastdbのオプションは以下の通り。
|
1 2 3 4 5 6 7 8 9 |
USAGE makeblastdb [-h] [-help] [-in input_file] [-input_type type] -dbtype molecule_type [-title database_title] [-parse_seqids] [-hash_index] [-mask_data mask_data_files] [-mask_id mask_algo_ids] [-mask_desc mask_algo_descriptions] [-gi_mask] [-gi_mask_name gi_based_mask_names] [-out database_name] [-blastdb_version version] [-max_file_sz number_of_bytes] [-metadata_output_prefix ] [-logfile File_Name] [-taxid TaxID] [-taxid_map TaxIDMapFile] [-version] |
makeblastdbの実行。
|
1 |
makeblastdb -in ./02ref/MiFishDB.fasta -input_type fasta -dbtype nucl -title 'MiFishDB' -logfile ./03log/makeblastdb.log -out ./02ref/MiFishDB |
02refフォルダの中には以下の8ファイルが生成されていればOKです。
・MiFishDB.ndb
・MiFishDB.nhr
・MiFishDB.nin
・MiFishDB.njs
・MiFishDB.not
・MiFishDB.nsq
・MiFishDB.ntf
・MiFishDB.nto
次にBlastnを実行します。
オプションは以下の通り。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 |
USAGE blastn [-h] [-help] [-import_search_strategy filename] [-export_search_strategy filename] [-task task_name] [-db database_name] [-dbsize num_letters] [-gilist filename] [-seqidlist filename] [-negative_gilist filename] [-negative_seqidlist filename] [-taxids taxids] [-negative_taxids taxids] [-taxidlist filename] [-negative_taxidlist filename] [-entrez_query entrez_query] [-db_soft_mask filtering_algorithm] [-db_hard_mask filtering_algorithm] [-subject subject_input_file] [-subject_loc range] [-query input_file] [-out output_file] [-evalue evalue] [-word_size int_value] [-gapopen open_penalty] [-gapextend extend_penalty] [-perc_identity float_value] [-qcov_hsp_perc float_value] [-max_hsps int_value] [-xdrop_ungap float_value] [-xdrop_gap float_value] [-xdrop_gap_final float_value] [-searchsp int_value] [-sum_stats bool_value] [-penalty penalty] [-reward reward] [-no_greedy] [-min_raw_gapped_score int_value] [-template_type type] [-template_length int_value] [-dust DUST_options] [-filtering_db filtering_database] [-window_masker_taxid window_masker_taxid] [-window_masker_db window_masker_db] [-soft_masking soft_masking] [-ungapped] [-culling_limit int_value] [-best_hit_overhang float_value] [-best_hit_score_edge float_value] [-subject_besthit] [-window_size int_value] [-off_diagonal_range int_value] [-use_index boolean] [-index_name string] [-lcase_masking] [-query_loc range] [-strand strand] [-parse_deflines] [-outfmt format] [-show_gis] [-num_descriptions int_value] [-num_alignments int_value] [-line_length line_length] [-html] [-sorthits sort_hits] [-sorthsps sort_hsps] [-max_target_seqs num_sequences] [-num_threads int_value] [-mt_mode int_value] [-remote] [-version] |
実行するコードは以下の通り。
|
1 2 3 4 5 6 7 8 9 10 |
blastn \ -query ./01output/other/Representative_seq.fasta \ -db ./02ref/MiFishDB \ -evalue 1e-10 \ -num_threads 24 \ -perc_identity 90 \ -qcov_hsp_perc 95 \ -max_target_seqs 100 \ -out ./01output/other/blastres.tsv \ -outfmt "6 qseqid sseqid length mismatch gapopen qstart qend sstart send evalue bitscore qseq" |
色々と指定しているオプションは以下の通りです(NCBI)。
| オプション | 説明 |
| query | 相同性検索したい配列データ |
| db | 検索対象となるデータベース |
| evalue | E-value |
| length | query配列に対するデータベース配列のアライメント長の閾値 |
| num_threads | blast検索時に使用するthread数 |
| perc_identity | 配列相同性の閾値 |
| qcov_hsp_perc | query配列に対してデータベース配列が重複している割合の閾値 |
| bitscore | Bit score |
| max_target_seqs | 結果を出力する最大配列数 |
| outfmt | 6を指定するとタブ区切り形式で結果を出力(7はコメントありタブ区切り形式) |
出力形式を指定するoutfmtに使用した値に関する情報は以下のとおりです。
| オプション | 説明 |
| qseqid | クエリ配列の名前 |
| sseqid | データベース配列の名前 |
| length | アライメント配列長 |
| mismatcah | データベース配列に対するクエリ配列のミスマッチ数 |
| gapopen | データベース配列に対するクエリ配列のギャップ数 |
| qstart | クエリ配列のアライメント開始位置 |
| qend | クエリ配列のアライメント終了位置 |
| sstart | データベース配列のアライメント開始位置 |
| send | データベース配列のアライメント終了位置 |
| evalue | E-value |
| bitscore | Bit score |
| qseq | クエリ配列 |
出力された結果ファイルはタブ区切りテキストなのでエクセルなどを使って確認することが出来ます。
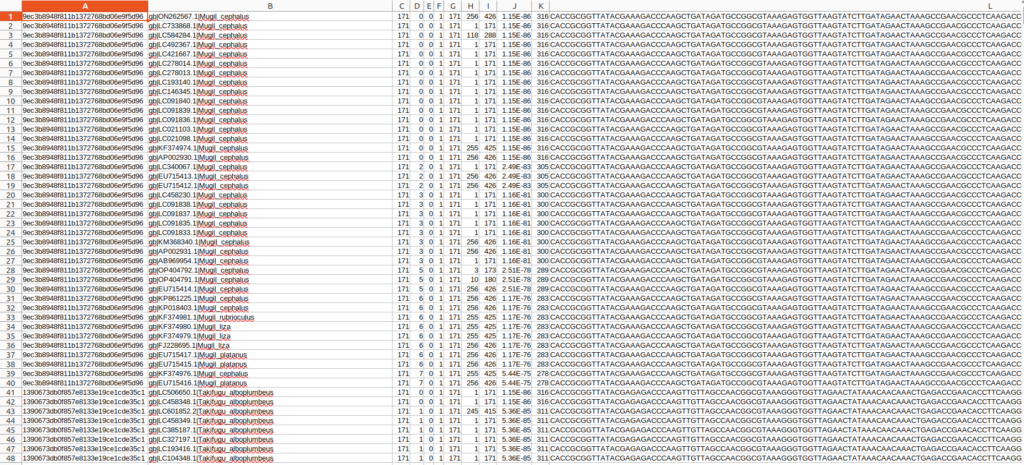
参照したデータベースの型が悪く、sseqid(2列目)の形式が微妙なので整形します。
gb|の削除とアクセッション番号の列の生成。
|
1 |
sed -E 's/gb\|//g; s/\|/\t/g' ./01output/other/blastres.tsv > ./01output/other/blastres_sep.tsv |
出力結果の1行目にoutmftで指定した情報を基本としたヘッダーを作成
|
1 |
sed -e '1i Query_seqid\tAcc\tScientific_name\tLength\tMismatch\tGapopen\tQstart\tQend\tSstart\tSend\tE-value\tBitscore\tQseq' ./01output/other/blastres_sep.tsv > ./01output/other/blastres_sep_hd.tsv |
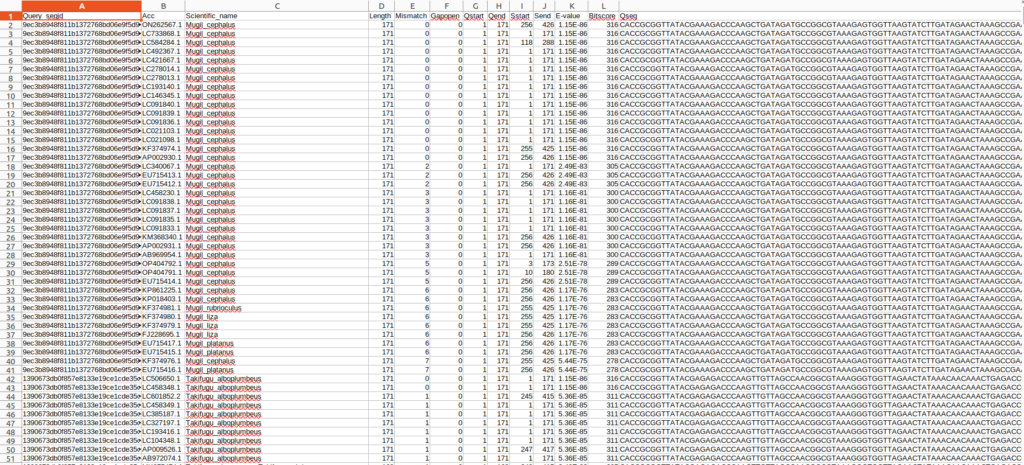
これで、クエリ配列に対してBlast検索で最大100本までの検索結果の情報を得ることが出来ました。ただ、このままでは同じ種に関する情報が重複していて非常に扱いづらいです。
クエリ配列に対して、Scientific name, Length, Mismatch, Gapopen, E-value, Bitscoreが重複している場合、重複を除去することでスッキリします(データベースがキュレーションされている前提)。
また、出力されているBlastの結果はBit scoreに基づいてSortされており、今回の場合であれば上にある情報ほどクエリ配列の種である可能性が高いです。なので、重複する情報を削除をすることで上位〇〇の配列情報を抽出できるはずです。
今回の結果だと、Queray_seqid:9ec3b8948f811b1372768bd06e9f5d96に対して検索結果が最大100配列分記載されていますが、色のついている部分のMugil cephalusについては、100%一致で情報が重複しています。正しく同定された個体に基づく配列なのであれば、どのAccession numberの情報でもMugil cephalusには変わなさそうです。
なのでこの場合、どれか1本の配列情報に代表させてしまってもMugil cephalusが100%一致でHitしたという事実には変わらないでしょう。以降のMismatchが2や3となっている場合や別種についても同様です。
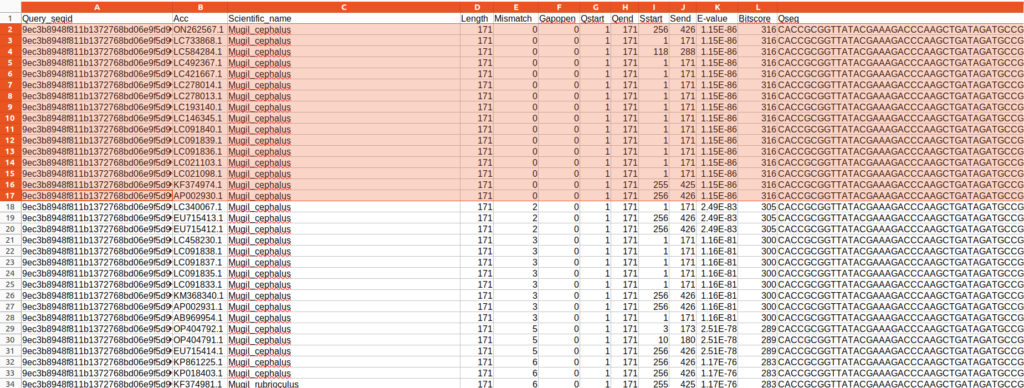
以降は、私がbashスクリプトで処理しきれないためpythonベースでデータ処理していきます。
Blastnで出力された結果から上位10の配列情報に整形する
スクリプト&記事作成中…
まとめ
- Miniconda3を使ってQIIME2をインストールしました
- SRA toolkitを使ってハイスループットシーケンスデータの取得を実施しました
- QIIME2を用いてハイスループットシーケンスデータの解析を実施しました
- BLAST+をインストールしてQIIME2で得られた配列データに対して相同性検索を実施しました
以降、追記予定
- 相同性検索の結果、検出された配列に対してその由来である可能性のある種の情報 上位10個をテーブルデータに反映させます。
備考
ダウンロードしたfastqファイルのNNNNNNの数の不一致について
fastq.gzファイルのプライマーのNNNNNN数の不一致について、以下はseqfuのviewオプションを使ってMiFish-UnityをDRR321580_R1.fastq.gzに対して検索かけた結果です。
|
1 2 |
seqfu view ./00fastqgz/DRR321580_R1.fastq.gz -o GYYGGTAAAWCTCGTGCCAGC --match-ths 1 seqfu view ./00fastqgz/DRR321580_R2.fastq.gz -o CAAACKRGGATTAGAYACCCYMCTATG --match-ths 1 |
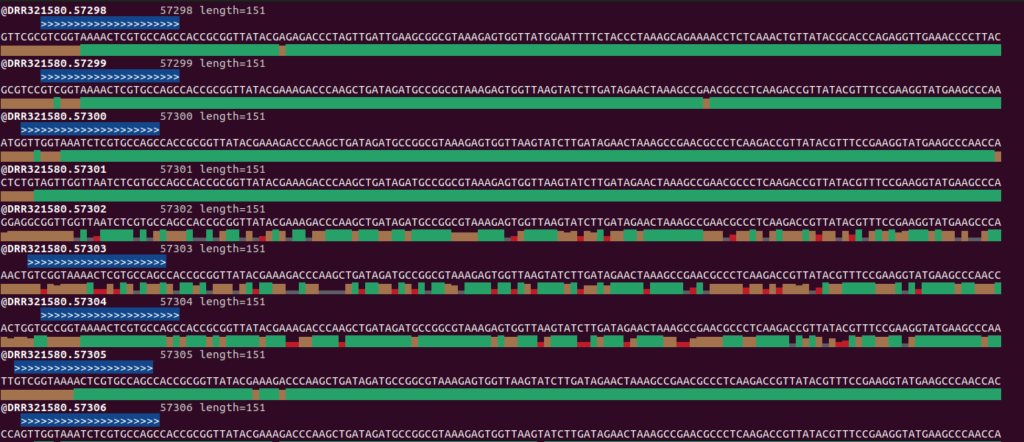

>>>はMiFish-Unity-F, <<<はMiFish-Unity-Rを表しています。それより左はNNNNNNに該当する部分ですが、2塩基のときもあれば6塩基のときもあるといった感じです。使用するプライマーのNの数を変えることは配列決定の際、同じ配列が多いようなライブラリでも識別しやすくなるので、そういったことをしているのかもしれません。ただ、プライマー除去時に<NNNNNN + プライマー配列長>だとプライマー配列が残ったり識別領域の配列を削除してしまったりするので今回はCutadaptを使ってプライマー配列を除去しました。
また、DADA2のDocumentationにも配列長を指定して削除するのはプライマーが読み取りの開始点にあり、長さが一定である場合であり、最終的にプライマー配列はしっかりと除去しておくようにとのコメントがあります。
How can I remove primers/adapters/etc from my amplicon reads?
If primers are at the start of your reads and are a constant length (a common scenario) you can use the
trimLeft = c(FWD_PRIMER_LEN, REV_PRIMER_LEN)argument of dada2’s filtering functions to remove the primers.For more complex situations (e.g. heterogenity spacers, variable length amplicons such as the ITS region) we recommend the cutadapt tool or the trimmomatic tool. You can see a worked example, including verification of primer removal, in the ITS-specific version of the DADA2 workflow.
Please double-check that your primers have been removed! The ambiguous nucleotides in unremoved primers will interfere with the dada2 pipeline.
https://benjjneb.github.io/dada2/faq.html
ここらへんはデータ処理の配列処理の注意点なので生データ等をしっかり見て処理を進めたいですね。
BLASTn -outfmtのオプション一覧
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 |
alignment view options: 0 = pairwise, 1 = query-anchored showing identities, 2 = query-anchored no identities, 3 = flat query-anchored, show identities, 4 = flat query-anchored, no identities, 5 = XML Blast output, 6 = tabular, 7 = tabular with comment lines, 8 = Text ASN.1, 9 = Binary ASN.1 10 = Comma-separated values 11 = BLAST archive format (ASN.1) 12 = Seqalign (JSON), 13 = Multiple-file BLAST JSON, 14 = Multiple-file BLAST XML2, 15 = Single-file BLAST JSON, 16 = Single-file BLAST XML2, 17 = Sequence Alignment/Map (SAM), 18 = Organism Report Options 6, 7, and 10 can be additionally configured to produce a custom format specified by space delimited format specifiers. The supported format specifiers are: qseqid : Query Seq-id qgi : Query GI qacc : Query accesion sseqid : Subject Seq-id sallseqid : All subject Seq-id(s), separated by a ';' sgi : Subject GI sallgi : All subject GIs sacc : Subject accession sallacc : All subject accessions qstart : Start of alignment in query qend : End of alignment in query sstart : Start of alignment in subject send : End of alignment in subject qseq : Aligned part of query sequence sseq : Aligned part of subject sequence evalue : Expect value bitscore : Bit score score : Raw score length : Alignment length pident : Percentage of identical matches nident : Number of identical matches mismatch : Number of mismatches positive : Number of positive-scoring matches gapopen : Number of gap openings gaps : Total number of gap ppos : Percentage of positive-scoring matches frames : Query and subject frames separated by a '/' qframe : Query frame sframe : Subject frame btop : Blast traceback operations (BTOP) staxids : unique Subject Taxonomy ID(s), separated by a ';'(in numerical order) sscinames : unique Subject Scientific Name(s), separated by a ';' scomnames : unique Subject Common Name(s), separated by a ';' sblastnames : unique Subject Blast Name(s), separated by a ';' (in alphabetical order) sskingdoms : unique Subject Super Kingdom(s), separated by a ';' (in alphabetical order) stitle : Subject Title salltitles : All Subject Title(s), separated by a '<>' sstrand : Subject Strand qcovs : Query Coverage Per Subject (for all HSPs) qcovhsp : Query Coverage Per HSP qcovus : measure of Query Coverage that counts a position in a subject sequence for this measure only once. The second time the position is aligned to the query is not counted towards this measure. When not provided, the default value is: 'qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore', which is equivalent to the keyword 'std' |
Ref
- SeqFu: A Suite of Utilities for the Robust and Reproducible Manipulation of Sequence Files
https://www.mdpi.com/2306-5354/8/5/59 - Qiime2 を用いた 16S rRNA 菌叢解析
https://qiita.com/keisuke-ota/items/6399b2f2f7459cd9e418 - QIIME2 forum
https://forum.qiime2.org/ - QIIME2
https://qiime2.org/
免責事項
本記事の情報・内容については可能な限り十分注意は払っていますが、正確性や完全性、妥当性および公平性について保証するものではありません。本記事の利用や閲覧によって生じたいかなる損害について責任を負いません。また、本記事の情報は予告なく変更される場合がありますので、ご理解くださいますようお願い申し上げます。





