ちょうど面白い検討も兼ねて、Miseqをランする機会でもあったので、お題の通り環境DNAの技術を流用して種の同定にチャレンジしました。@しばた
対象は普段苔の下に隠れているこいつです。上司が毎日採ってきてくれるダンゴムシが主食です。

仕事の疲れを癒してくれる可愛いペットですが、種類が正確に分かりません(外見上、採種してもいい種だと検討はついていますが自分の同定能力にはあまり自信がありません)。
では、外見で種類が分からないならやっぱりDNAを読んでみるしかないでしょう。
方法 – サンプリングからシーケンスまで –
普通個体が目の前にいたら組織片を採種して、DNeasyなどでDNA抽出するかと思います。
ですが、大切なペットなので可能な限り傷つけたくありません。野外で絶滅危惧種を見つけた時と同じ気持ちです。
そこで非侵襲性に長けている環境DNAの技術使います。
お題にも“環境DNAの技術を使って”と書いているので、いつもと同じようにフィルターを使います。

やる事は簡単で、このフィルターでサンショウウオをなでまわします。目的は体表面の”ヌメリ”です。
水をろ過して種を検出できるなら、ヌメリからなら余裕でしょう。
このフィルターをTsuji et al.2020に従って、DNA抽出します。

そんなに使わないので、最終溶出量を50μLで溶出しました。
私がいる会社には、サンガーシーケンサーはないので今回はMiseqを使います。Miseqのカートリッジの最大配列長は600bpなので、それより短い増幅産物長でサンショウウオを見分けられるようなプライマーを使います。
以前、ヤマトサンショウウオを野外の水から検出するのに使ったこの論文のプライマーを使いました。

種を見分ける領域は290bpなのでMiseqでも可読出来る長さです。
論文の通りに1stPCR, 2ndPCRとライブラリ調整を進めて、E-gelで泳動して精製ました。

バイオアナライザーで長さを確認した後に、かなり薄く希釈してMiseqで配列を読みました。
方法 – 解析 –
Miseqから出力されるデータは基本的にFastqというファイル形式になっています。もっと大元から解析する人はbcl(バイナリベースコール形式)ファイルを使ったりします。
大型脊椎動物などを対象とした環境DNA分析の場合、このFastqファイルから生物の種名とおおよそのDNA配列の量を示すRead数が記載されたSummary Tableを取得するのに使う解析法はUsearchとDADA2にBlastを組み合わせた方法が主流だと思います。
両手法とも確か、Amplicon Sequence Variantを略してASV(ESVと呼ばれる場合もあります)といった、OTU(Operational Taxonomic Unit)より解像度を上げたレベルで配列処理を行ってたと思います。
今回はRと呼ばれる解析ソフトウェアで簡単に動かすことの出来るDADA2とweb上で行えるBlast検索によってFastqファイルからMiseqで読まれた配列を取り出して、エラー配列除去などの処理をしたのちに、種を同定していきました。
実際にRをインストールしてDADA2で解析して、出てきた配列からWebのBlastで検索して、多様性を検出する。というのもそのうち記事で書こうと思っているので、お待ちください。(最近忙しい)
とりあえず進めていきます。
2本鎖のDNAをそれぞれを一部重複するようにMiseqで読んでいるので、DADA2では配列のクオリティプロットを見て、どの程度の長さを残して重複部分で結合(Merge)させるかを指定します。
フォワード側よりリバース側の方が後半にクオリティが下がりやすいので多めに削ることが多いです。
プライマーの配列を削除したり、error rateの推定やキメラの除去を経た後に出力された配列データがこれです(思ったより濃い、、)。
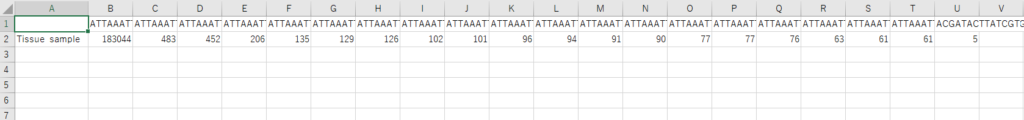
この配列データをfasta形式にします。
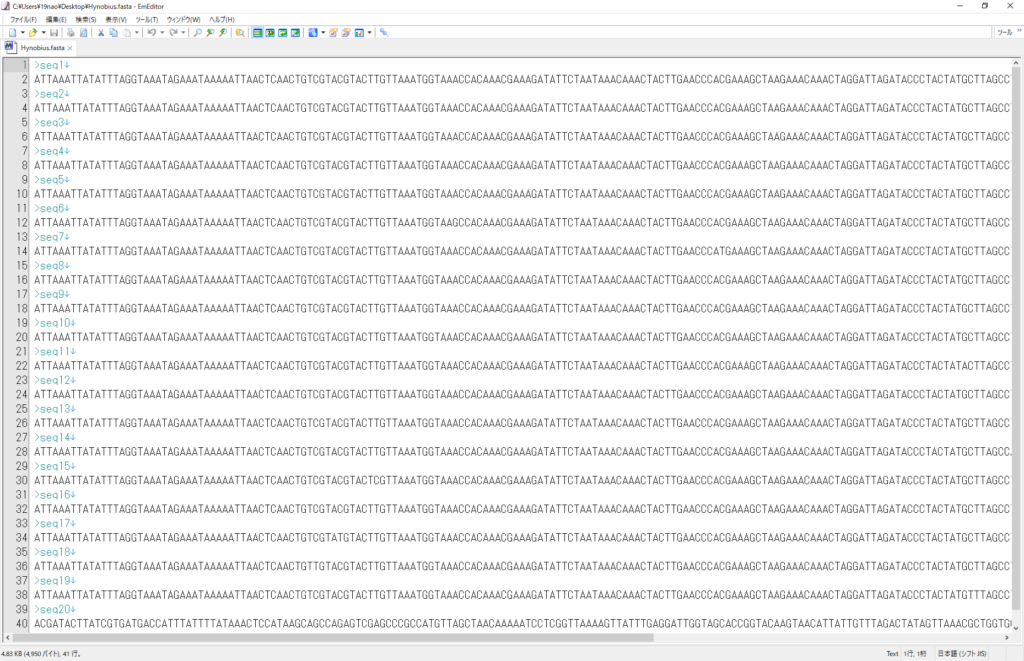
webのblast検索にかけます。
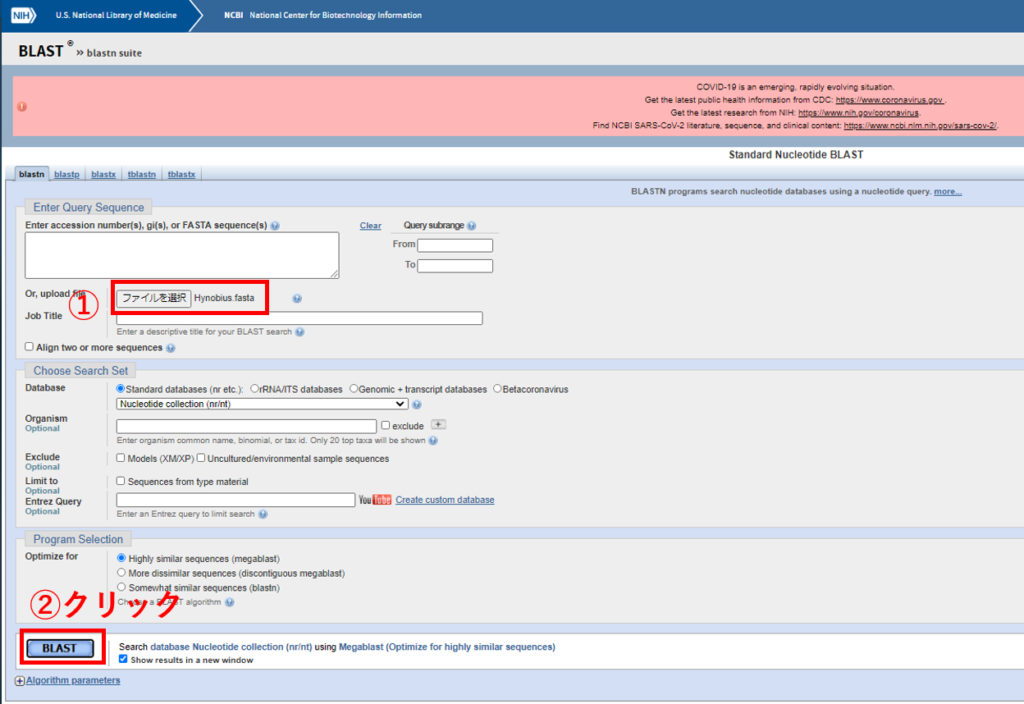
Enter accession number(s) …の部分に普通の環境DNA分析で分析会社からもらった代表配列をコピペして入れてみると、その配列はどの種のものと近いのか見ることも出来ます。
結果
最もRead数の多かった配列をBlast検索した結果がこちらです。
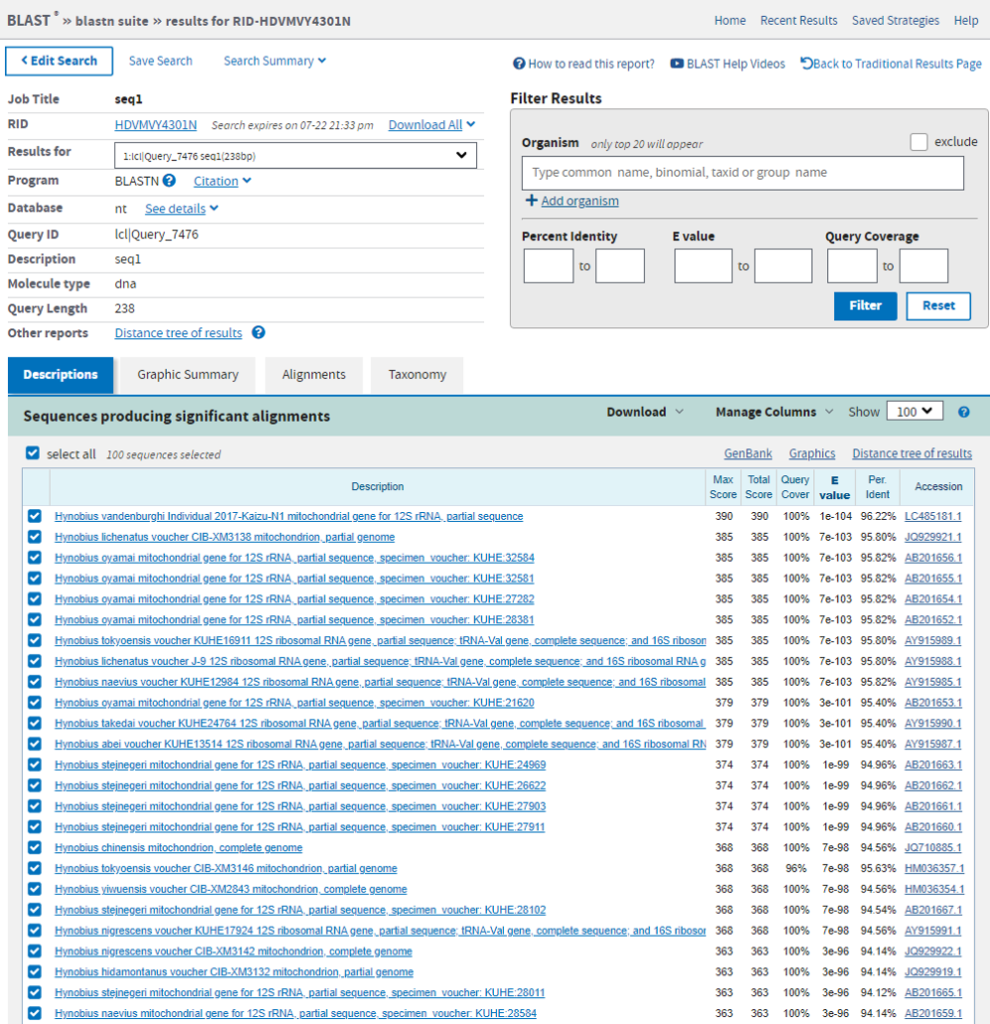
Miseqで読んだ配列と96.22%の一致率(9塩基違い)でHynobius vandenburghib ヤマトサンショウウオでした。

ダントツでRead数が多かった配列とその他の配列との関係性を見るためにMega7で樹形図を書いてみたところ、各配列はダントツで多かったseq1と非常に類似する配列でした。もしかしたら、Miseqの読み間違いやPCR時の増幅ミスによるエラー配列の可能性があります。

ダントツでRead数が多かったseq1以外もseq1と数塩基しか変わらないので、Miseqの読み間違いやPCRの増幅ミスによって生成された配列の可能性。
また、細胞内には複数のミトコンドリアDNAがあるので1個体でも複数のハプロタイプを持っている場合がありますが、今回はハプロタイプの検出ではなく種を同定したいので、最も多いRead数の配列を採用すればいいと考えました。また、個体が持つ真の配列より残ったエラーなどの偽の配列数が多くなることは理論上無いはずですので、最もRead数が多い配列を選択することは間違ってないかと思います。
総括?
今回は環境DNAで使う技術で非侵襲的に個体を同定しようとしました。ほぼ、個体を傷つけずに配列を読むことには成功したのですが、HopHitの種と9塩基も違うとなると、多分これはプライマー領域の部分の配列が登録されていないヤマトサンショウウオではない種の可能性が高いです。ただ厳密にHynobius属の遺伝距離から考察しているわけではないので、憶測には過ぎません。
次にとる方法としては、Matsui et al.2020に従ってサンガーシーケンスするのが、DNAから種を同定する最適な方法かな思います。
もし、データベースが充実していて、私の思っている種が登録されていれば、Miseqを使った種の同定もできたかもしれません。
環境DNAでこの個体が生息している流域で水を採水して分析した場合も、TopHitと9塩基も違うとTopHitの種です!という結論も出せないので、やはりデータベースの拡充は重要な課題であると言えます。
生物分類ができる人がいてこそ、環境DNAなどのDNAをベースとした技術の能力が最大限に発揮されるので、私個人としては微力ながらどうにか分類学者を応援したいところです(そして形態ベースで私のペットを誰か同定してください)。
最後まで読んでいただきありがとうございました。