MitoFish とMiFishパイプライン: 環境DNAメタバーコーディングのための分析パイプラインを備えた魚類のミトコンドリアゲノムデータベース
MitoAnnotatorを実装しているMitoFishの機能的アップデートに関する報告論文です。中心的な内容は魚類のミトコンドリアの環境DNA分析のためのMiFishパイプラインの実装について記載されています。

この解析パイプラインはデータを処理するうえで上図Aのような複数のソフトウェアが組み合わされて構築されています。
使い方は簡単です。 サイト内の説明通りに進めていきましょう。
サムネイル画像のGet started nowのボタンを押せばこの画面に移ります
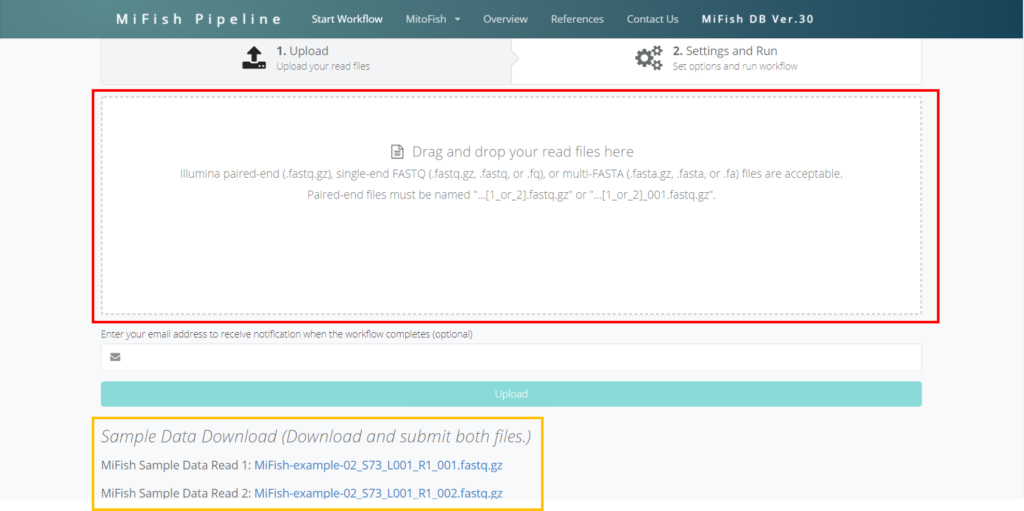
次世代シークエンサーから出力されたFastq.gzもしくはFastqファイルを持っている方はドラッグ&ドロップで赤枠のにポイっと入れてください。
もし、Fastqファイルを持っていない方は、オレンジ枠のFastq.gzファイルをそれぞれダウンロードしてから、同様に赤枠の部分にポイっと入れてください。

ポイっと入れたらUploadしましょう。
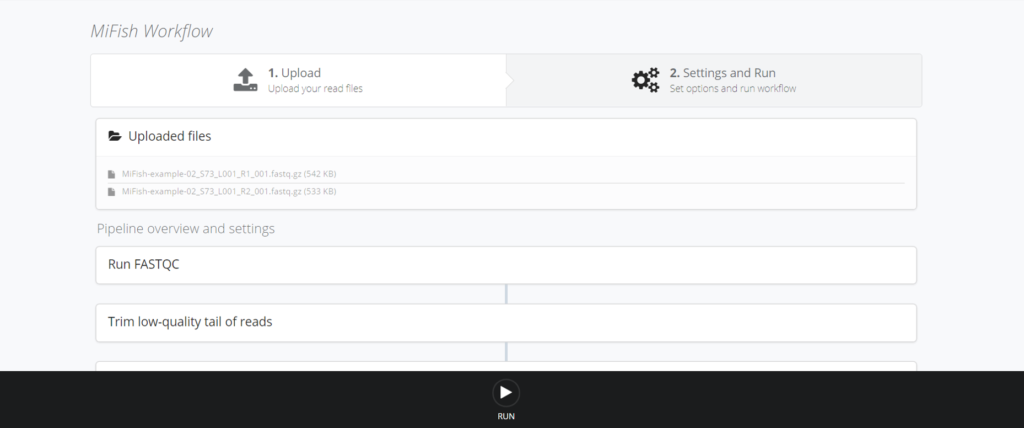
アップロードが完了するとこんな画面に移行します。下に変更可能な設定値がいくつかあります。
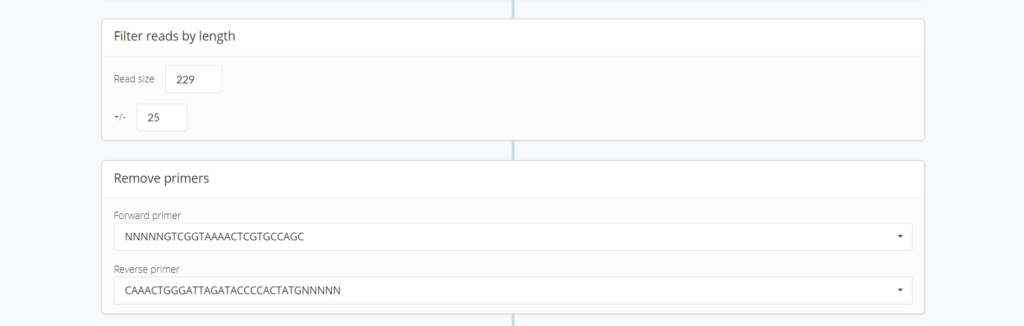
Filter reads by length : フィルタリングする配列長と除去するプライマー配列を設定します。
Remove primer : プライマーの配列を変更する部分だと思いますが今は変更できないようです。
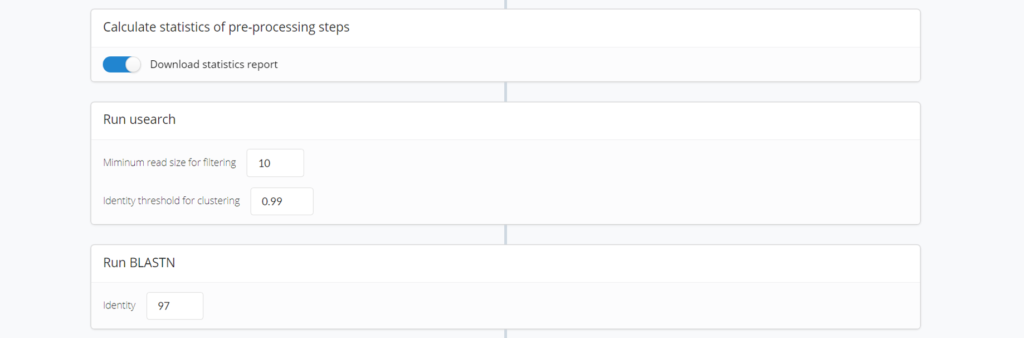
Download statistics report : 解析が終わった後にレポートを出力するかどうか
Minimum read size for filtering : 出力の際の最低リード数はどうするのか
Identity threshold for clustering : 1塩基, 2塩基違いでDNA配列が混在するなかで、1つの代表となる配列を何塩基違いまでまとめるか
Run BLAST(Identity) : Miseqで解読された配列でフィルタリング後に残った代表配列との相同性検索の際にトップヒットを何パーセントまで許容するかなどが設定できます。
基本的に設定は変えなくていいと思います。
それでは下のRunのボタンを押して解析を開始しましょう。
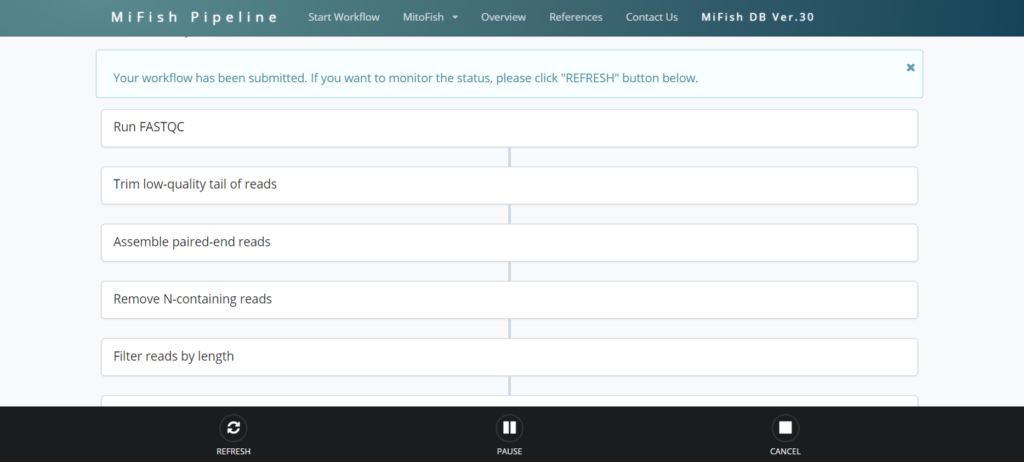
解析を開始すると現状の処理がどこまで進んでいるのか確認することが出来ます。
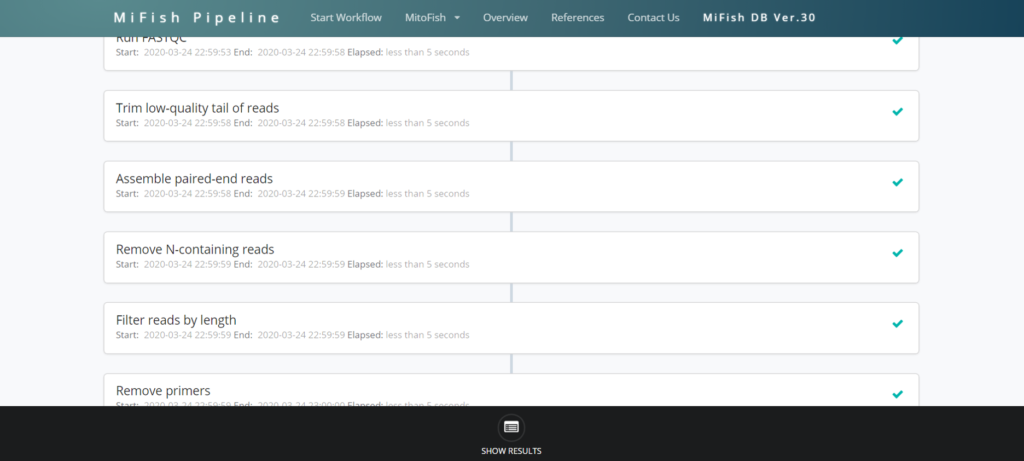
全ての処理工程が終われば、Show Resultsで解析結果を確認することが出来ます。
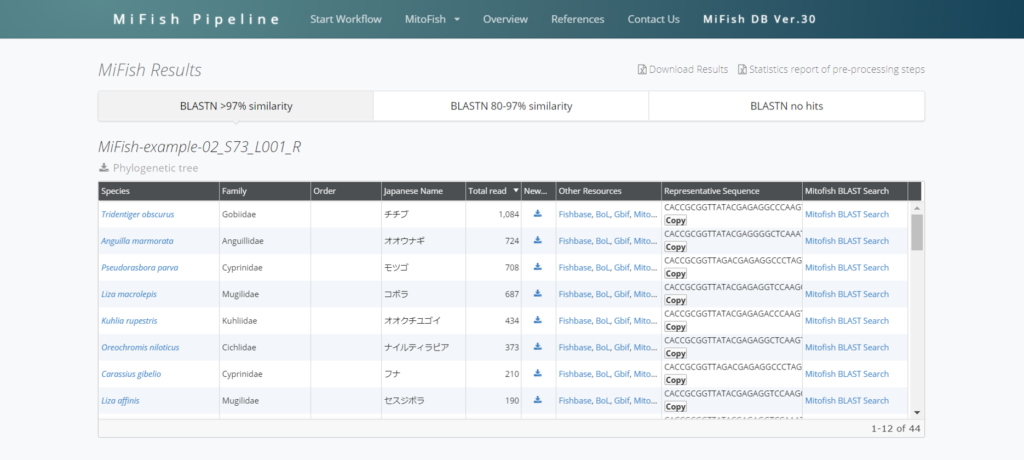
オオクチユゴイやオオウナギなどが検出されているので、このサンプルは沖縄でしょうか。
Representative sequenceはその種であると決めるもととなった代表配列のことです。
右上のDownload Resultsで結果を丸ごとダウンロードすることが出来ます。
ここではWebのMiFish pipelineを紹介しましたが、このパイプラインを使わなくてもMiFishを用いたメタバーコーディング法の解析が可能です。論文とデモデータを用いた解説をこちらでしております。勉強しながら書いているので少し解釈に間違いがあるかもしれませんが、1度目を通してもらえると知見が広まるかと思います。@しばた
画像はジャーナルのCC BYに従う形で、記事で紹介したオープンアクセスの論文中の画像または、自身で作成したものを使用しています。