
この記事の内容
きっかけ
NCBIのFTPサイトで提供されている塩基配列データは以下のような形式になっています。
>NR_025636.1 Mycobacterium shottsii strain M175 16S ribosomal RNA, partial sequence
GTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCTTAACACATGCAAGTCGAACGGAAAGGTCTCCTCGGAGATACT…..
>NR_025639.1 Aequorivita antarctica strain SW49 16S ribosomal RNA, partial sequence
GAACGCTAGCGGCAGGCCTAACACATGCAAGTCGAGGGGTAGAGAGTGCTTGCGCTCTTGAGACCGGCGCACGGGTGCGT…..
一方、Microbiomeにおけるmetagenomicsの参照データベースとして一般的に用いられる、SILVAやGreengeneは以下のように複数の分類階級を記載する形式となっています。
>(識別番号1)
配列ファイル (.fasta)
GTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCTTAACACATGCAAGTCGAACGGAAAGGTCTCCTCGGAGATACT….
>(識別番号2)
GAACGCTAGCGGCAGGCCTAACACATGCAAGTCGAGGGGTAGAGAGTGCTTGCGCTCTTGAGACCGGCGCACGGGTGCGT…..
(識別番号1) k__Bacteria;p__Actinomycetota;c__Actinomycetes;o__Corynebacteriales;f__Mycobacteriaceae;g__Mycobacterium;s__Mycobacterium shottsii
(識別番号2) k__Bacteria;p__Bacteroidota;c__Flavobacteriia;o__Flavobacteriales;f__Flavobacteriaceae;g__Aequorivita;s__Aequorivita antarctica
分類情報ファイル (.tsv)
NCBI Refseqは種や属といった単一階級の情報しか無いため、少し使用しづらいなと感じていました。
もう少し使いやすいように、NCBI Accession Numberをキーとして、SILVAなどで採用されている複数の分類階級を記載する形式に変換してみよう。というのが今回の内容になります。
注意点として、データベースのキュレーションの方法がSILVA, Greengenes, NCBI-RefSeq, GTDB, RDPで異なり、どのデータベースを使用するかにより、分類の解像度や結果が異なる可能性があります(ref1, ref2)。Metabarcodingなどでデータベースとして利用する際にはそれぞれの特徴を理解した上で使用してください。
今回使用するのはNCBI Refseq 16s rRNAの配列データとNCBI taxonomy databaseのデータセットです。
全体のながれ
流れは以下の通りです。
- NCBI ftp siteよりRefseqのbacteria 16s rRNAのBlast DBをダウンロード
- blastdbcmdにてBlast DB形式からfastaへ
- Taxonkit, csvtkを使って、単一階級から複数階級の情報を持つtsv ファイルと、Accession numberで紐付くfastaファイルを作成 (QIIME2ではセットで使います)
先にまとめコードを示しておきます。
本記事で説明する内容のうち、”配列データの取得”以降の作業がまとめられてます。
- デスクトップ上似作成したbioinfoフォルダ下にq2reformatというフォルダを作成
- その中にqiime2-reformat.shというファイルを作成して以下のまとめコードを記載
- 本記事の”配列データの取得”の後まで実施した後に
bash qiime2-reformat.shを実行
そうすれば配列データの16S_ribosomal_RNA_seq_acc.fastaと分類情報が記載されている16S_ribosomal_RNA_tax_qiime2-form.tsvが生成されるはずです。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 |
#!/usr/bin/bash set -eu # variable out_qza="01output/qza" out_qzv="01output/qzv" out_other="01output/other" blast_res='01output/other/blast_res' ref="02ref" # make directory mkdir -p ./01output/qza ./01output/qzv ./01output/other ./01output/other/blast_res ./02ref # =================== makedb =================== # blastdbcmd -entry all -db ./${ref}/16S_ribosomal_RNA -dbtype nucl > \ ./${ref}/16S_ribosomal_RNA.fasta # import database time qiime tools import \ --input-path ./${ref}/16S_ribosomal_RNA.fasta \ --output-path ./${ref}/16S_ribosomal_RNA.qza \ --input-format "DNAFASTAFormat" \ --type "FeatureData[Sequence]" # === make folder === # mkdir ./${ref}/_temp # === make taxonomy file === # blastdbcmd -entry all \ -db ./${ref}/16S_ribosomal_RNA \ -dbtype nucl \ -outfmt "%T" | \ awk '!a[$0]++' | sort > ./${ref}/_temp/16S_ribosomal_RNA_tax blastdbcmd -entry all \ -db ./${ref}/16S_ribosomal_RNA \ -outfmt "%T##%a" | \ sed -E 's/##/\t/g' | sort > ./${ref}/_temp/16S_ribosomal_RNA_tax-acc # assign lineage to taxids taxonkit lineage -j 24 ./${ref}/_temp/16S_ribosomal_RNA_tax \ -o ./${ref}/_temp/16S_ribosomal_RNA_tax_taxid-lineage.tsv # Reformat lineages taxonkit reformat ./${ref}/_temp/16S_ribosomal_RNA_tax_taxid-lineage.tsv \ -j 24 --output-ambiguous-result -F -P | \ cut -f 1,3 > ./${ref}/_temp/16S_ribosomal_RNA_tax_taxid-lineage_qiime2_form.tsv # separate taxid with no classification information awk '{ if ($2 == "") print $0}' \ ./${ref}/_temp/16S_ribosomal_RNA_tax_taxid-lineage_qiime2_form.tsv \ > ./${ref}/_temp/16S_ribosomal_RNA_tax_taxonomy_no_lineage.tsv awk '{ if ($2 != "") print $0}' \ ./${ref}/_temp/16S_ribosomal_RNA_tax_taxid-lineage_qiime2_form.tsv \ > ./${ref}/_temp/16S_ribosomal_RNA_tax_taxonomy.tsv # concat qiime2 form taxonomic information if [ $(wc -l < "./${ref}/_temp/16S_ribosomal_RNA_tax_taxonomy_no_lineage.tsv") -eq 0 ]; then csvtk -t -l join -f 1 --left-join -H \ ./${ref}/_temp/16S_ribosomal_RNA_tax-acc ./${ref}/_temp/16S_ribosomal_RNA_tax_taxonomy.tsv | \ cut -f 2,3 > ./${ref}/_temp/16S_ribosomal_RNA_tax_qiime2-form else csvtk -t -l join -f 1 --left-join -H \ ./${ref}/_temp/16S_ribosomal_RNA_tax-acc ./${ref}/_temp/16S_ribosomal_RNA_tax_taxonomy.tsv \ cut -f 2,3 > ./${ref}/_temp/16S_ribosomal_RNA_tax_qiime2-form1 csvtk -t -l join -f 1 --left-join -H \ ./${ref}/_temp/16S_ribosomal_RNA_tax-acc ./${ref}/_temp/16S_ribosomal_RNA_tax_taxonomy_no_lineage.tsv \ --na 'k__NoInfo;p__;c__;o__;f__;g__;s__' | \ cut -f 2,3 > ./${ref}/_temp/16S_ribosomal_RNA_tax_qiime2-form2 cat ./${ref}/_temp/16S_ribosomal_RNA_tax_qiime2-form1 ./${ref}/_temp/16S_ribosomal_RNA_tax_qiime2-form2 > ./${ref}/_temp/16S_ribosomal_RNA_tax_qiime2-form fi # add header in 16S_ribosomal_RNA_tax_qiime2-form.tsv sed '1i Feature ID\tTaxon' ./${ref}/_temp/16S_ribosomal_RNA_tax_qiime2-form > \ ./${ref}/16S_ribosomal_RNA_tax_qiime2-form.tsv # output fasta blastdbcmd -entry all -db ./${ref}/16S_ribosomal_RNA \ -dbtype nucl -outfmt ">%a##%s" | \ sed -E 's/##/\n/g' > \ ./${ref}/16S_ribosomal_RNA_seq_acc.fasta # remove temp file rm -r ./${ref}/_temp/ |
内容
準備
作業ディレクトリの準備
q2reformatというフォルダをデスクトップのbioinfoというフォルダ内に作成します。デスクトップ上でコマンドラインを開いて以下のように入力。
|
1 |
mkdir -p ./bioinfo/q2reformat && cd ./bioinfo/q2reformat |
次に出力フォルダ等を作成
|
1 |
mkdir -p ./02ref ./02ref/_temp |
移行はq2reformatをベースに02refと_tempフォルダにデータをどんどん出力していきます。
Taxonokit, csvtk, blastdbcmdのインストール
Taxonkit
Taxonkitは、NCBI Taxonomy Databaseから取得した分類情報を含むtaxonomy dump fileを取り扱うためのコマンドラインツールです。taxonomy dump fileを読み込んで、分類群の分類階級、学名、シノニム、分類群の識別子などの情報を取得し、変換することができます。

開発者githubはこちら。オプションは以下の通り。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 |
TaxonKit - A Practical and Efficient NCBI Taxonomy Toolkit Version: 0.14.1 Author: Wei Shen <shenwei356@gmail.com> Source code: https://github.com/shenwei356/taxonkit Documents : https://bioinf.shenwei.me/taxonkit Citation : https://www.sciencedirect.com/science/article/pii/S1673852721000837 Dataset: Please download and uncompress "taxdump.tar.gz": ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz and copy "names.dmp", "nodes.dmp", "delnodes.dmp" and "merged.dmp" to data directory: "/home/naoki/.taxonkit" or some other directory, and later you can refer to using flag --data-dir, or environment variable TAXONKIT_DB. When environment variable TAXONKIT_DB is set, explicitly setting --data-dir will overide the value of TAXONKIT_DB. Usage: taxonkit [command] Available Commands: cami-filter Remove taxa of given TaxIds and their descendants in CAMI metagenomic profile create-taxdump Create NCBI-style taxdump files for custom taxonomy, e.g., GTDB and ICTV filter Filter TaxIds by taxonomic rank range genautocomplete generate shell autocompletion script (bash|zsh|fish|powershell) lca Compute lowest common ancestor (LCA) for TaxIds lineage Query taxonomic lineage of given TaxIds list List taxonomic subtrees of given TaxIds name2taxid Convert scientific names to TaxIds profile2cami Convert metagenomic profile table to CAMI format reformat Reformat lineage in canonical ranks taxid-changelog Create TaxId changelog from dump archives version print version information and check for update Flags: --data-dir string directory containing nodes.dmp and names.dmp (default "/home/naoki/.taxonkit") -h, --help help for taxonkit --line-buffered use line buffering on output, i.e., immediately writing to stdin/file for every line of output -o, --out-file string out file ("-" for stdout, suffix .gz for gzipped out) (default "-") -j, --threads int number of CPUs. 4 is enough (default 4) --verbose print verbose information Use "taxonkit [command] --help" for more information about a command. |
taxdumpファイルのダウンロード
NCBIのFPTサイトよりtaxdump.tar.gzファイルをダウンロードして、ワークディレクトリに保存用フォルダを作成後、必要なファイルを移動させておきます。
|
1 2 3 4 |
wget ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz && tar -zxvf taxdump.tar.gz mkdir -p $HOME/.taxonkit cp names.dmp nodes.dmp delnodes.dmp merged.dmp $HOME/.taxonkit rm *.dmp *.prt taxdump.tar.gz |
最新の分類情報を使用したい場合、結構な頻度で更新かかってるので、こまめにNCBIのTaxonomy dumpファイルを更新して常に最新のものにすることをおすすめします。
- FPT site : https://ftp.ncbi.nih.gov/pub/taxonomy/
Taxonokitのインストール
作業する仮想環境あればactivateして以下を実行します。
|
1 |
mamba install -c bioconda taxonokit -y |
仮想環境がない場合は、作成してインストール
|
1 2 |
mamba create -n q2reformat taxonkit -y mamba activate q2reformat |
taxonkit -hでhelpが表示されればインストール完了
csvtk
csvtkはカンマ区切り、またはタブ区切りテキストをコマンドライン上で編集するツールです。似たものにcsvkitがありますが、csvtkの方が後発なため包括関係になってます。また、awkより簡潔なので使いやすいです。
| Features | csvtk | csvkit | Note |
|---|---|---|---|
| Read Gzip | Yes | Yes | read gzip files |
| Fields ranges | Yes | Yes | e.g. -f 1-4,6 |
| Unselect fileds | Yes | — | e.g. -1 for excluding first column |
| Fuzzy fields | Yes | — | e.g. ab* for columns with name prefix “ab” |
| Reorder fields | Yes | Yes | it means -f 1,2 is different from -f 2,1 |
| Rename columns | Yes | — | rename with new name(s) or from existed names |
| Sort by multiple keys | Yes | Yes | bash sort like operations |
| Sort by number | Yes | — | e.g. -k 1:n |
| Multiple sort | Yes | — | e.g. -k 2:r -k 1:nr |
| Pretty output | Yes | Yes | convert CSV to readable aligned table |
| Unique data | Yes | — | unique data of selected fields |
| frequency | Yes | — | frequencies of selected fields |
| Sampling | Yes | — | sampling by proportion |
| Mutate fields | Yes | — | create new columns from selected fields |
| Replace | Yes | — | replace data of selected fields |
開発者githubはこちら。オプションは以下の通り。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 |
csvtk -- a cross-platform, efficient and practical CSV/TSV toolkit Version: 0.25.0 Author: Wei Shen <shenwei356@gmail.com> Documents : http://shenwei356.github.io/csvtk Source code: https://github.com/shenwei356/csvtk Attention: 1. The CSV parser requires all the lines have same number of fields/columns. Even lines with spaces will cause error. Use '-I/--ignore-illegal-row' to skip these lines if neccessary. 2. By default, csvtk thinks your files have header row, if not, switch flag "-H" on. 3. Column names better be unique. 4. By default, lines starting with "#" will be ignored, if the header row starts with "#", please assign flag "-C" another rare symbol, e.g. '$'. 5. By default, csvtk handles CSV files, use flag "-t" for tab-delimited files. 6. If double quotes exist in fields, use flag "-l". 7. Do not mix use field (column) numbers and names. Environment variables for frequently used global flags: - "CSVTK_T" for flag "-t/--tabs" - "CSVTK_H" for flag "-H/--no-header-row" You can also create a soft link named "tsvtk" for "csvtk", which sets "-t/--tabs" by default. Usage: csvtk [command] Available Commands: add-header add column names cat stream file to stdout and report progress on stderr comb compute combinations of items at every row concat concatenate CSV/TSV files by rows corr calculate Pearson correlation between two columns csv2json convert CSV to JSON format csv2md convert CSV to markdown format csv2rst convert CSV to reStructuredText format csv2tab convert CSV to tabular format csv2xlsx convert CSV/TSV files to XLSX file cut select and arrange fields del-header delete column names dim dimensions of CSV file filter filter rows by values of selected fields with arithmetic expression filter2 filter rows by awk-like arithmetic/string expressions fmtdate format date of selected fields fold fold multiple values of a field into cells of groups freq frequencies of selected fields gather gather columns into key-value pairs genautocomplete generate shell autocompletion script (bash|zsh|fish|powershell) grep grep data by selected fields with patterns/regular expressions head print first N records headers print headers inter intersection of multiple files join join files by selected fields (inner, left and outer join) mutate create new column from selected fields by regular expression mutate2 create new column from selected fields by awk-like arithmetic/string expressions ncol print number of columns nrow print number of records plot plot common figures pretty convert CSV to readable aligned table rename rename column names with new names rename2 rename column names by regular expression replace replace data of selected fields by regular expression round round float to n decimal places sample sampling by proportion sep separate column into multiple columns sort sort by selected fields space2tab convert space delimited format to CSV split split CSV/TSV into multiple files according to column values splitxlsx split XLSX sheet into multiple sheets according to column values summary summary statistics of selected numeric or text fields (groupby group fields) tab2csv convert tabular format to CSV transpose transpose CSV data unfold unfold multiple values in cells of a field uniq unique data without sorting version print version information and check for update watch monitor the specified fields xlsx2csv convert XLSX to CSV format Flags: -c, --chunk-size int chunk size of CSV reader (default 50) -C, --comment-char string lines starting with commment-character will be ignored. if your header row starts with '#', please assign "-C" another rare symbol, e.g. '$' (default "#") -d, --delimiter string delimiting character of the input CSV file (default ",") -h, --help help for csvtk -E, --ignore-empty-row ignore empty rows -I, --ignore-illegal-row ignore illegal rows --infile-list string file of input files list (one file per line), if given, they are appended to files from cli arguments -l, --lazy-quotes if given, a quote may appear in an unquoted field and a non-doubled quote may appear in a quoted field -H, --no-header-row specifies that the input CSV file does not have header row -j, --num-cpus int number of CPUs to use (default value depends on your computer) (default 24) -D, --out-delimiter string delimiting character of the output CSV file, e.g., -D $'\t' for tab (default ",") -o, --out-file string out file ("-" for stdout, suffix .gz for gzipped out) (default "-") -T, --out-tabs specifies that the output is delimited with tabs. Overrides "-D" -t, --tabs specifies that the input CSV file is delimited with tabs. Overrides "-d" Use "csvtk [command] --help" for more information about a command. |
csvtkのインストール
作業する仮想環境あればそちらにactivateして以下を実行します。
|
1 |
mamba install -c bioconda csvtk -y |
blastdbcmd
NCBI BLASTの一部で、BLAST DB内のシーケンス情報などを抽出することができます。他にもたくさんの機能はありますが、ここでは必要な情報の抽出程度にしか使用しません。
オプションは以下の通り。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 |
USAGE blastdbcmd [-h] [-help] [-db dbname] [-dbtype molecule_type] [-entry sequence_identifier] [-entry_batch input_file] [-ipg IPG] [-ipg_batch input_file] [-taxids taxonomy_ids] [-taxidlist input_file] [-info] [-metadata] [-metadata_output_prefix ] [-tax_info] [-range numbers] [-strand strand] [-mask_sequence_with mask_algo_id] [-out output_file] [-outfmt format] [-target_only] [-get_dups] [-line_length number] [-ctrl_a] [-show_blastdb_search_path] [-list directory] [-remove_redundant_dbs] [-recursive] [-list_outfmt format] [-exact_length] [-long_seqids] [-logfile File_Name] [-version] DESCRIPTION BLAST database client, version 2.13.0+ Use '-help' to print detailed descriptions of command line arguments |
blastdbcmdのインストール
blastdbcmdはBLAST+に内包されています。そのため、BLAST+をインストールすれば使用できるようになります。こちらの記事でも解説しましたが、mambaを使って導入可能です。
|
1 |
mamba install -c bioconda 'blast>=2.13' -y |
完了したら確認。
|
1 |
blastdbcmd -h |
並列でファイルを回答可能なpigzもインストール
|
1 |
mamaba install -c conda-forge pigz -y |
配列データの取得
NCBI FTPサイトよりBLAST DB形式になっているデータを取得します。
|
1 |
wget ftp://ftp.ncbi.nlm.nih.gov/blast/db/16S_ribosomal_RNA.tar.gz -P ./02ref/ && tar -I pigz -xvf ./02ref/16S_ribosomal_RNA.tar.gz |
.tarファイルを解凍して以下のファイルが出力されていればOK。
|
1 |
tree 02ref/ |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 |
02ref/ ├── 16S_ribosomal_RNA.ndb ├── 16S_ribosomal_RNA.nhr ├── 16S_ribosomal_RNA.nin ├── 16S_ribosomal_RNA.nnd ├── 16S_ribosomal_RNA.nni ├── 16S_ribosomal_RNA.nog ├── 16S_ribosomal_RNA.nos ├── 16S_ribosomal_RNA.not ├── 16S_ribosomal_RNA.nsq ├── 16S_ribosomal_RNA.ntf ├── 16S_ribosomal_RNA.nto ├── 16S_ribosomal_RNA.tar.gz ├── taxdb.btd └── taxdb.bti |
次にblastdbcmdを使って、BLAST DB形式から配列データなどを抽出します。
配列データ
multifastaファイルが得られます。
|
1 |
blastdbcmd -entry all -db ./02ref/16S_ribosomal_RNA -dbtype nucl > ./02ref/16S_ribosomal_RNA.fasta |
中身を確認してみます。
|
1 |
cat ./02ref/16S_ribosomal_RNA.fasta | head |
taxid, taxidとAccession numberの対応表の取得
Taxonkitで使用するtaxidは以下のように取得します。
|
1 |
blastdbcmd -entry all -db ./02ref/16S_ribosomal_RNA -dbtype nucl -outfmt "%T" | awk '!a[$0]++' | sort > ./02ref/_temp/16S_ribosomal_RNA_tax |
スクリプトで実施しているのは以下になります。
- blastdbcmdの
outfmtに%Tを使用することでBLAST DBに登録されている配列のtaxidを抽出 - awkで重複の削除 & sortで並び替え
出力ファイルの内容をcatでチェック
|
1 |
cat ./02ref/_temp/16S_ribosomal_RNA_tax | head |
|
1 2 3 4 5 6 7 8 9 10 |
100 1000562 1000565 1000566 1000568 1000570 1001 1001240 100133 100134 |
うまく得られているようです。
次は、taxidとAccession Numberとの対応表を作成します。
|
1 |
blastdbcmd -entry all -db ./02ref/16S_ribosomal_RNA -outfmt "%T##%a" | sed -E 's/##/\t/g' | sort > ./02ref/_temp/16S_ribosomal_RNA_tax-acc |
スクリプトで実施しているのは以下になります。
- blastdbcmdの
outfmtに%Tに加えて%aを指定することでaccession numberを抽出(区切り文字は一時的に##とした) - sedで##をタブに変換 & sort
Taxonkitを使ってtaxidに系統分類の情報を付与する
ここでTaxonkitが登場します。taxonkitのlineageオプションを使用することで、taxonomy dumpファイルにtaxidで検索をかけて系統分類の情報を抽出していきます (-j オプションは自身のPCのコアを指定してください)
|
1 |
taxonkit lineage -j 24 ./02ref/_temp/16S_ribosomal_RNA_tax -o ./02ref/_temp/16S_ribosomal_RNA_tax_taxid-lineage.tsv |
ここで[WARN]と表示されることがあります。2023/4/2現在だと以下のような表示でした。
|
1 2 3 |
21:29:04.050 [WARN] taxid 1176194 was merged into 2878561 21:29:04.053 [WARN] taxid 1841867 was merged into 2897707 21:29:04.054 [WARN] taxid 2582907 was merged into 2729629 |
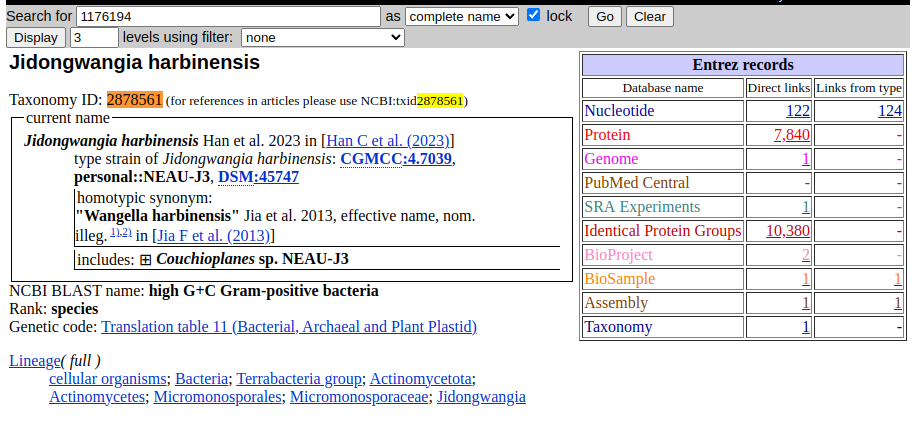
試しにNCBI taxonomyで対象のtaxidとして1176194を調べてみると、merged into 2878561と表示されていたように、Hitしたのは2878561に割り振られている生物の情報でした。配列が登録された後に分類が見直されたりとかしたのですかね。
このような変更は多々あるので、taxonomy dumpファイルは可能な限り最新のものかversionが分かるように管理して利用することが望ましいと思います。
出力したファイルを見てみましょう。
|
1 |
cat ./02ref/_temp/16S_ribosomal_RNA_tax_taxid-lineage.tsv | head |
|
1 2 3 4 5 6 7 8 9 10 |
100 cellular organisms;Bacteria;Pseudomonadota;Alphaproteobacteria;Hyphomicrobiales;Xanthobacteraceae;Ancylobacter;Ancylobacter aquaticus 1000562 cellular organisms;Bacteria;Terrabacteria group;Bacillota;Bacilli;Lactobacillales;Streptococcaceae;Streptococcus;Streptococcus phocae;Streptococcus phocae subsp. salmonis 1000565 cellular organisms;Bacteria;Pseudomonadota;Betaproteobacteria;Nitrosomonadales;Sterolibacteriaceae;Methyloversatilis;Methyloversatilis universalis;Methyloversatilis universalis FAM5 1000566 cellular organisms;Bacteria;Terrabacteria group;Actinomycetota;Actinomycetes;Pseudonocardiales;Pseudonocardiaceae;Halosaccharopolyspora;Halosaccharopolyspora lacisalsi 1000568 cellular organisms;Bacteria;Terrabacteria group;Bacillota;Negativicutes;Veillonellales;Veillonellaceae;Megasphaera;Megasphaera lornae 1000570 cellular organisms;Bacteria;Terrabacteria group;Bacillota;Bacilli;Lactobacillales;Streptococcaceae;Streptococcus;Streptococcus anginosus group;Streptococcus anginosus;Streptococcus anginosus SK52 = DSM 20563 1001 cellular organisms;Bacteria;FCB group;Bacteroidetes/Chlorobi group;Bacteroidota;Cytophagia;Cytophagales;Bernardetiaceae;Garritya;Garritya polymorpha 1001240 cellular organisms;Bacteria;Terrabacteria group;Actinomycetota;Actinomycetes;Micrococcales;Microbacteriaceae;Cryobacterium;Cryobacterium roopkundense 100133 cellular organisms;Bacteria;Terrabacteria group;Bacillota;Clostridia;Eubacteriales;Lachnospiraceae;Sporobacterium;Sporobacterium olearium 100134 cellular organisms;Bacteria;Terrabacteria group;Bacillota;Clostridia;Eubacteriales;Lachnospiraceae;Anaerocolumna;Anaerocolumna xylanovorans |
先程抽出したtaxidに系統分類情報を付与することができました。
次はformatの修正です。taxonkitのreformatオプションを使用して、k__;p__;c__;o__;f__;__;g__;s__の形に変更します。
|
1 |
taxonkit reformat ./02ref/_temp/16S_ribosomal_RNA_tax_taxid-lineage.tsv -j 24 -a -F -P | cut -f 1,3 > ./02ref/_temp/16S_ribosomal_RNA_tax_taxid-lineage_qiime2_form.tsv |
出力したファイルを見てみましょう。
|
1 |
cat ./02ref/_temp/16S_ribosomal_RNA_tax_taxid-lineage_qiime2_form.tsv | head |
|
1 2 3 4 5 6 7 8 9 10 |
100 k__Bacteria;p__Pseudomonadota;c__Alphaproteobacteria;o__Hyphomicrobiales;f__Xanthobacteraceae;g__Ancylobacter;s__Ancylobacter aquaticus 1000562 k__Bacteria;p__Bacillota;c__Bacilli;o__Lactobacillales;f__Streptococcaceae;g__Streptococcus;s__Streptococcus phocae 1000565 k__Bacteria;p__Pseudomonadota;c__Betaproteobacteria;o__Nitrosomonadales;f__Sterolibacteriaceae;g__Methyloversatilis;s__Methyloversatilis universalis 1000566 k__Bacteria;p__Actinomycetota;c__Actinomycetes;o__Pseudonocardiales;f__Pseudonocardiaceae;g__Halosaccharopolyspora;s__Halosaccharopolyspora lacisalsi 1000568 k__Bacteria;p__Bacillota;c__Negativicutes;o__Veillonellales;f__Veillonellaceae;g__Megasphaera;s__Megasphaera lornae 1000570 k__Bacteria;p__Bacillota;c__Bacilli;o__Lactobacillales;f__Streptococcaceae;g__Streptococcus;s__Streptococcus anginosus 1001 k__Bacteria;p__Bacteroidota;c__Cytophagia;o__Cytophagales;f__Bernardetiaceae;g__Garritya;s__Garritya polymorpha 1001240 k__Bacteria;p__Actinomycetota;c__Actinomycetes;o__Micrococcales;f__Microbacteriaceae;g__Cryobacterium;s__Cryobacterium roopkundense 100133 k__Bacteria;p__Bacillota;c__Clostridia;o__Eubacteriales;f__Lachnospiraceae;g__Sporobacterium;s__Sporobacterium olearium 100134 k__Bacteria;p__Bacillota;c__Clostridia;o__Eubacteriales;f__Lachnospiraceae;g__Anaerocolumna;s__Anaerocolumna xylanovorans |
Reformatすることができました。
もう少し修正
Refseqに登録されているデータはキュレーションされたものなので良いのですが、ntデータベースを対象に種名を利用してlineageのアサイメントをすると、分類情報が登録されていない情報が出てきます。
その中で全くlineageに関する情報が振られていないものは、振られていない = “NoInfo”と情報つけておく方が後々助かるのでcsvtkを使って情報の付与を行います。
|
1 2 3 4 5 6 |
awk '{ if ($2 == "") print $0}' \ ./02ref/_temp/16S_ribosomal_RNA_tax_taxid-lineage_qiime2_form.tsv \ > ./02ref/_temp/16S_ribosomal_RNA_tax_taxonomy_no_lineage.tsv awk '{ if ($2 != "") print $0}' \ ./02ref/_temp/16S_ribosomal_RNA_tax_taxid-lineage_qiime2_form.tsv \ > ./02ref/_temp/16S_ribosomal_RNA_tax_taxonomy.tsv |
今回の場合、16S_ribosomal_RNA_tax_taxonomy_no_lineage.tsvのデータ量は0 byteになりますが、ifで条件分岐させてエラーが出ないようにしました。
以下の作業で出力される16S_ribosomal_RNA_tax_taxonomy.tsvは先ほど確認した、Reformat後のデータと同じです。
分割していたファイルを結合する処理が以下になります。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 |
if [ $(wc -l < "./02ref/_temp/16S_ribosomal_RNA_tax_taxonomy_no_lineage.tsv") -eq 0 ]; then csvtk -t -l join -f 1 --left-join -H \ ./02ref/_temp/16S_ribosomal_RNA_tax-acc ./02ref/_temp/16S_ribosomal_RNA_tax_taxonomy.tsv | \ cut -f 2,3 > ./02ref/_temp/16S_ribosomal_RNA_tax_qiime2-form else csvtk -t -l join -f 1 --left-join -H \ ./02ref/_temp/16S_ribosomal_RNA_tax-acc ./02ref/_temp/16S_ribosomal_RNA_tax_taxonomy.tsv \ cut -f 2,3 > ./02ref/_temp/16S_ribosomal_RNA_tax_qiime2-form1 csvtk -t -l join -f 1 --left-join -H \ ./02ref/_temp/16S_ribosomal_RNA_tax-acc ./02ref/_temp/16S_ribosomal_RNA_tax_taxonomy_no_lineage.tsv \ --na 'k__NoInfo;p__;c__;o__;f__;g__;s__' | \ cut -f 2,3 > ./02ref/_temp/16S_ribosomal_RNA_tax_qiime2-form2 cat ./02ref/_temp/16S_ribosomal_RNA_tax_qiime2-form1 ./02ref/_temp/16S_ribosomal_RNA_tax_qiime2-form2 > ./02ref/_temp/16S_ribosomal_RNA_tax_qiime2-form fi |
後々、QIIME2で使用することを考慮して”Feature ID”と”Taxon”というheaderを追加します。
|
1 2 |
sed '1i Feature ID\tTaxon' ./02ref/_temp/16S_ribosomal_RNA_tax_qiime2-form > \ ./02ref/16S_ribosomal_RNA_tax_qiime2-form.tsv |
出力したファイルを見てみましょう。
|
1 |
cat ./02ref/16S_ribosomal_RNA_tax_qiime2-form.tsv | head |
|
1 2 3 4 5 6 7 8 9 10 |
Feature ID Taxon NR_133868.1 k__Bacteria;p__Bacillota;c__Bacilli;o__Lactobacillales;f__Streptococcaceae;g__Streptococcus;s__Streptococcus phocae NR_043813.1 k__Bacteria;p__Pseudomonadota;c__Betaproteobacteria;o__Nitrosomonadales;f__Sterolibacteriaceae;g__Methyloversatilis;s__Methyloversatilis universalis NR_118131.1 k__Bacteria;p__Actinomycetota;c__Actinomycetes;o__Pseudonocardiales;f__Pseudonocardiaceae;g__Halosaccharopolyspora;s__Halosaccharopolyspora lacisalsi NR_181404.1 k__Bacteria;p__Bacillota;c__Negativicutes;o__Veillonellales;f__Veillonellaceae;g__Megasphaera;s__Megasphaera lornae NR_041722.2 k__Bacteria;p__Bacillota;c__Bacilli;o__Lactobacillales;f__Streptococcaceae;g__Streptococcus;s__Streptococcus anginosus NR_112705.1 k__Bacteria;p__Bacillota;c__Bacilli;o__Lactobacillales;f__Streptococcaceae;g__Streptococcus;s__Streptococcus anginosus NR_117426.1 k__Bacteria;p__Bacillota;c__Bacilli;o__Lactobacillales;f__Streptococcaceae;g__Streptococcus;s__Streptococcus anginosus NR_104500.1 k__Bacteria;p__Actinomycetota;c__Actinomycetes;o__Micrococcales;f__Microbacteriaceae;g__Cryobacterium;s__Cryobacterium roopkundense NR_024967.1 k__Bacteria;p__Bacillota;c__Clostridia;o__Eubacteriales;f__Lachnospiraceae;g__Sporobacterium;s__Sporobacterium olearium |
無事、headerが追加されたようです。
Accession numberが付与されたfastaファイルを取得する
最後にblastdbcmdでAccession numberを配列のheaderとするfastaファイルを出力します。
|
1 |
blastdbcmd -entry all -db ./02ref/16S_ribosomal_RNA -dbtype nucl -outfmt ">%a##%s" | sed -E 's/##/\n/g' > ./02ref/16S_ribosomal_RNA_seq_acc.fasta |
出力したファイルを見てみましょう。
|
1 |
cat ./02ref/16S_ribosomal_RNA_seq_acc.fasta | head |
|
1 2 3 4 5 6 7 8 9 10 |
>NR_025636.1 GTTTGATCCTGGCTCAGGACGAACGCTGGCGGCGTGCTTAACACATGCAAGTCGAACGGAAA..... >NR_025639.1 GAACGCTAGCGGCAGGCCTAACACATGCAAGTCGAGGGGTAGAGAGTGCTTGCGCTCTTGA....... >NR_025640.1 GAACGCTAGCGGCAGGCCTAACACATGCAAGTCGAGGGGTAGAAAGTGCTTGCACTTCTG....... >NR_025641.1 GAACGCTAGCGGCAGGCCTAACACATGCAAGTCGAGGGGTAGAAAGTGCTTGCACTTTTGA..... >NR_025646.1 GCAATACGTCAGCGGCAGACGGGTGAGTAACGCGTGGGAACGTACCTTTTGGTTCGGAACA....... |
Accession Numberが情報となった配列データベースが得られました。
ちなみに、2つのファイルに情報が別れているのが嫌な場合、seqkitのfx2tabを使用するとタブ区切りの情報をもとにfastaの配列情報部分を置換してくれます。
ここまできたら、16S_ribosomal_RNA_seq_acc.fastaと16S_ribosomal_RNA_tax_qiime2-form.tsvを使って、
- classify-consensus-blast
でASV/OTUに対してtaxonomic assignmentを実施したり、
fit-classifier-naive-bayesで分類機を作成後、classify-sklearnclassify-consensus-vsearchclassify-hybrid-vsearch-sklearn
などで、ASV/OTUに対してtaxonomic assignmentを実施することができると思います。
また、qiime2ベースでBLASTを実施する場合、1coreでの実行似制限されているのですが、query配列を分割することで事実上の並列処理が可能です。またその記事を公開したら↓にリンク掲載したいと思います。
↓ 更新しました @2023/5/14

最後までご覧いただきありがとうございました。
Reference
- Shen, W., Ren, H., TaxonKit: a practical and efficient NCBI Taxonomy toolkit, Journal of Genetics and Genomics, https://doi.org/10.1016/j.jgg.2021.03.006
免責事項
本記事の情報・内容については可能な限り十分注意は払っていますが、正確性や完全性、妥当性および公平性について保証するものではありません。本記事の利用や閲覧によって生じたいかなる損害について責任を負いません。また、本記事の情報は予告なく変更される場合がありますので、ご理解くださいますようお願い申し上げます。




