PMiFish pipiline v2.4.1
Miya et al. 2020で使用されたASV法を採用した新しいMiFishパイプラインです。
2020年12月現在のWeb版MiFishパイプラインはOTU法がベースにされていますが、PMiFishはノイズ除去で利点のあるUnoise3を用いたASV法と、Blast+より高速にデータベースとの照合をして合致する配列を探すことの出来るusearch_globalを採用しています。
Githubにあるチュートリアルに補足を加えながら従って進めていきます。
はじめに
Windowsを使用されている場合は、WSL(Windows Subsystem for Linux)の有効化とMicrosoft StoreからUbuntuをインストールして下さい。
やり方は下記に記載しております。また、仮想マシンソフトウェアVirtualBoxをによるLinuxのインストールも随時追加予定です。
どこかで筆者が解析に使った覚えのあるものを引っ張り出してまとめているので、環境を構築しても無駄ではないはず?です。

注意というわけではありませんが、インストール・コマンドの実行する前に全体をさっと確認してから進めてみてください。(2021/3/20追記)
動作環境
- Windows 10 Home
- WSL (Windows subsystem for Linux)
- Ubuntu 20.04 LTS
- ActivePerl 5.24.3 (windowsでperlを動かすために使用)
表記ルール
コマンドラインでのコマンドの表記
以下のように$から始まるものはshellスクリプトで、#はコメント行です。コマンドをubuntuにコピペする場合は、$より後ろをコピーして右クリックでペーストします。
|
1 2 |
# ファイルやディレクトリの情報を表示するコマンド $ ls |
ubuntu上で上記の$を抜いたコードを実行してみてください。実行する今開いているフォルダ(カレントディレクトリ)内にあるファイルやフォルダが一覧で表示されるはずです。
出力結果
出力結果がないとうまくいっているのか分かりにくいです。ここでは可能な限り下記のような出力結果を掲載するようにします。

ではいきます。
Ubuntuを起動してgit cloneできるようにする
いきなりチュートリアルには載っていませんが、解析するための環境を整える作業になります。
解析パッケージはソースコードがGithubにまとめられていることが多いです。
ディレクトリの構成などを1からまねて動作環境を作成するのはミスの原因にもなりえるので、開発者が提供してくれているファイル構成を複製(=clone)するのが一番楽です。
Githubからクローニングする準備をしていきます。
まず、デスクトップのアドレスバーに ubuntu.exeと打ってEnterを押すとubuntuが起動します。ちなみに背景色は設定で変えられます。

Gitをインストールします。
|
1 2 |
# パスワードを聞かれるのでubuntuをインストールした際に設定したパスワードを記入 $ sudo apt install git |
Gitがインストール出来たら以下のコマンドを実行してMiFishというフォルダをcドライブに作成します。
|
1 2 3 4 5 6 |
# ディレクトリをcドライブに移動 $ cd /mnt/c # cドライブにMiFishというフォルダを作る $ mkdir MiFish # MiFishフォルダに移動 $ cd MiFish |

Git cloneする
ファイル構成を複製(=clone)します。
|
1 |
$ git clone https://github.com/rogotoh/PMiFish.git |
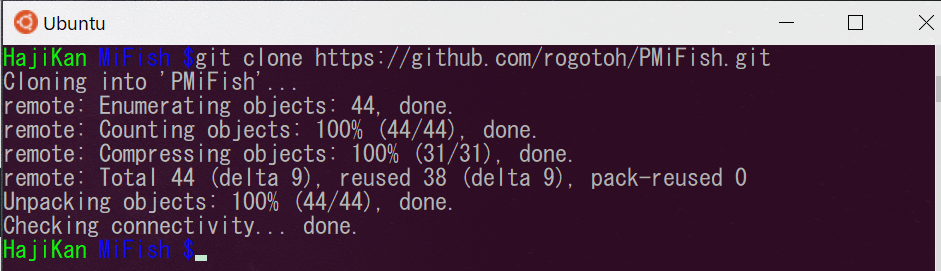
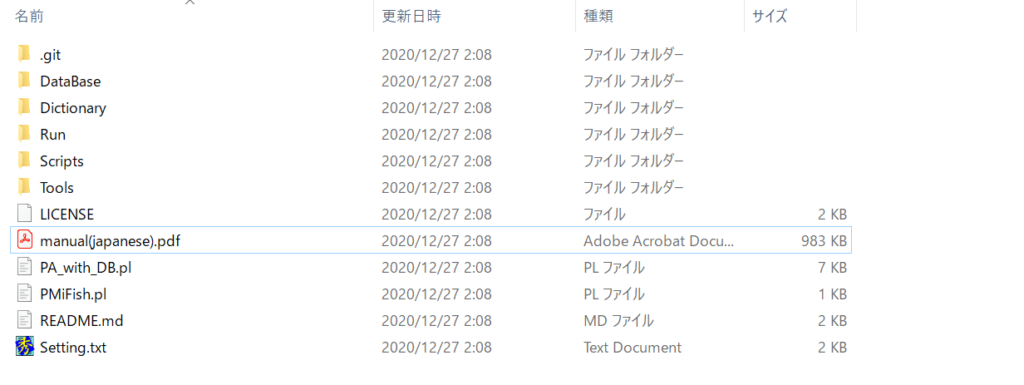
これでPMiFishを使うためのファイル一覧が手に入りました。
MiFishフォルダの中にできたPMiFishフォルダの中のmanual(japanese).pdfがPMiFish2.4.1の日本語マニュアルになります。記載されている指示に従っていただいても設定はできると思います。
使いそうな別パッケージもインストールしておく
ファイルをコマンドライン上からダウンロードするのに便利なwgetやファイルツリーを表示できるtreeなどインストールしておきます。
|
1 2 |
# パスワードを要求されるので記入する $ sudo apt install wget tree |
ActivePerl, Usearch11(, Gzip)を手に入れよう
ここからは必要な下記ソフトウェアをダウンロードしていきます。
- ActivePerl:Windowsでperlを動かすのに必要
https://www.activestate.com/activeperl - Usearch11:解析の中核を担うパッケージ群
https://www.drive5.com/usearch/download.html - Gzip:圧縮されているFastqファイルを解凍・圧縮する
http://gnuwin32.sourceforge.net/packages/gzip.htm
ActivePerlのインストール
https://www.activestate.com/products/perl/にアクセスすると下記画面が表示されます。
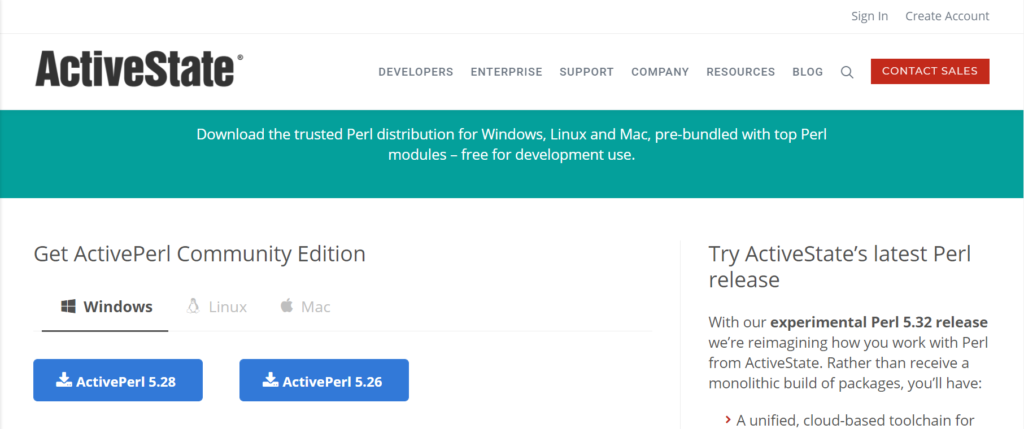
5.5Mbほどのイメージファイルがダウンロードフォルダにダウンロードされます。
Usearch11の実行ファイルをインストールしてパスを通す
マニュアルにはUsearhc11の実行ファイルをPMiFishフォルダ内にあるToolsフォルダに入れて、そこにパスを通すように。という指示があるのでそうします。
https://www.drive5.com/usearch/download.htmlにアクセスすると下記のような画面が表示されます。
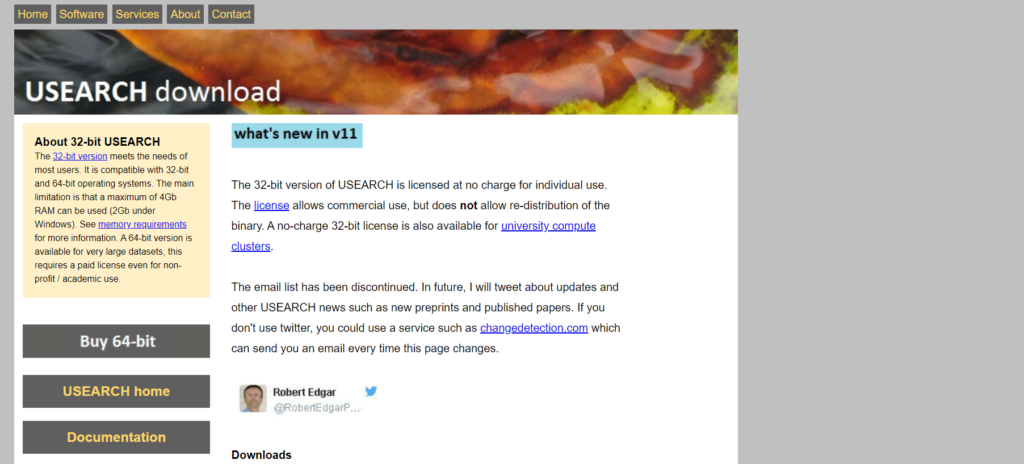
64bit版は有償なので今回は32bit版をダウンロードします。
ダウンロードされたgzファイルを解凍するのにフリーのソフトを使用します。
もし、フリーのソフトをダウンロードするのが嫌な場合は、2の手順でダウンロードとファイルの解凍を行ってください。
wgetというコマンドとPMiFishを動かす際に使用するGzipを使用して、ダウンロードと解凍をします。4コマンドくらいで終わるので楽です。
1 手動でダウンロードしてLhaplus(フリーウェア)を使って解凍する
まず、Lhaplusをここからダウンロードします。
右の窓の社からダウンロードからファイルをダウンロードして指示に従って、Lhaplusをダウンロードします。
Lhaplusをダウンロードし終わったら、Usearchのページに戻り、windows用のusearch11.0.667_win32.exe.gzファイル(770kb)があるのでダウンロードします。
多分何もダウンロード先に指定をしていなければ、ダウンロードフォルダに配置されるので、そこで、右クリック > プログラムから開く > 別のプログラムを選択 > その他のアプリ↓ > Lhaplusを選択して実行します。
もし、Lhaplusが候補に無ければ、このPCで別のアプリを探す を押してcドライブのProgram Files(x86)にあるLhaplusという名前のフォルダの中にある Lhaplus.exeをダブルクリックします。
デスクトップに解凍されたフォルダが出来上がるので、その中のusearch11.0.667_win32.exeをコピーして、cドライブ > MiFish > PMiFish > Toolsの中にペーストします。
前はメールアドレスなどを求められた気がするのですがv11からは無いみたいですね。
2 wgetとgzipを使ってダウンロードと解凍をする
コマンドでUsearchの実行ファイルをダウンロードして解凍します。
今、Ubuntu上ではPMiFishフォルダにいるので、toolsフォルダに移動します。
|
1 |
$ cd Tools |
ここで、wgetコマンドを使ってダウンロードします。
|
1 |
$ wget https://www.drive5.com/downloads/usearch11.0.667_win32.exe.gz |
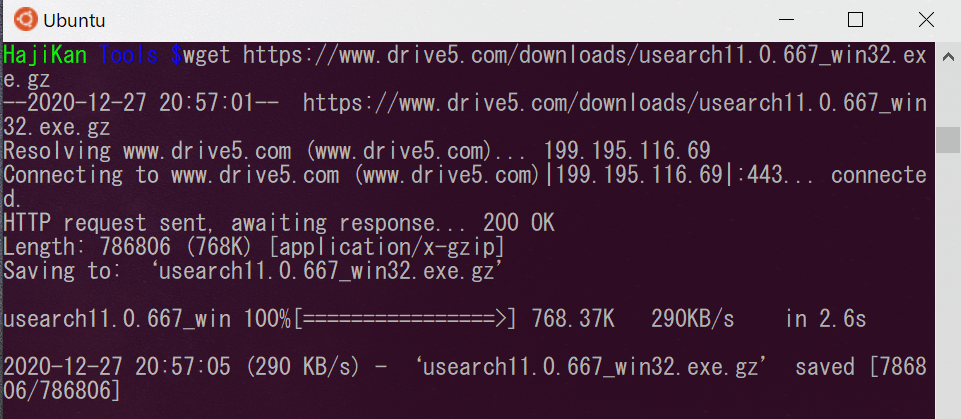
ダウンロードされたファイルがあるか確認します。
|
1 |
$ ls |

では、gzipを使って解凍します。
|
1 |
$ gzip -d usearch11.0.667_win32.exe.gz |
確認してみると.gzという拡張子が無くなったファイルがあると思います。これで、Usearchの実行ファイルのダウンロードと解凍は終わりです。
|
1 2 |
# 一つ前のフォルダに戻る $ cd .. |
Toolsフォルダでやる事が終わったので、一つ上の階層のフォルダに戻っておきます。
Usearch11の実行ファイルのパスを通す
PMiFishではToolsの中にUsearch11の実行ファイルがあるという前提でスクリプトが記述されているので、無い状態で実行すると実行ファイルをToolsにおいてくれとエラーがでます。
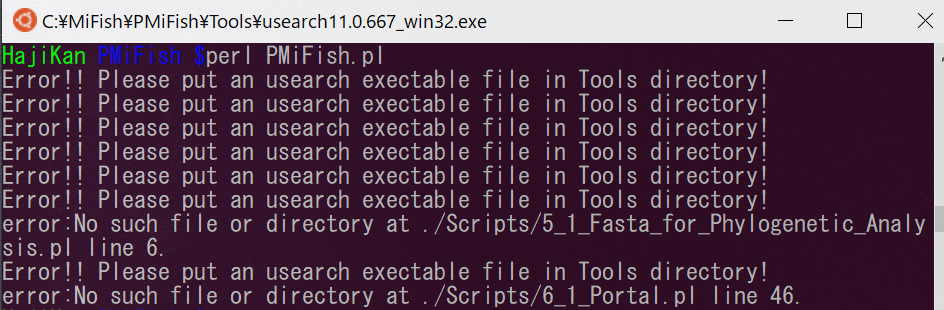
パスを通すとは何かというと、PCに動かしたいソフトの場所を教えてあげることだと思ってください。
Windowsでパスを通す簡単な方法は”ユーザー環境変数”に記載することです。ただ、ここには他のソフトのパスの設定もあるので、必要でなければ他の部分はいじらないでおきましょう。
やることは簡単で、他のパスの書き方に似せてUsearch11の実行ファイルのフォルダまでのパスを書き込むだけです。
まずWindowsのスタートメニューから設定を開いて、”システムの詳細設定”を開きます。
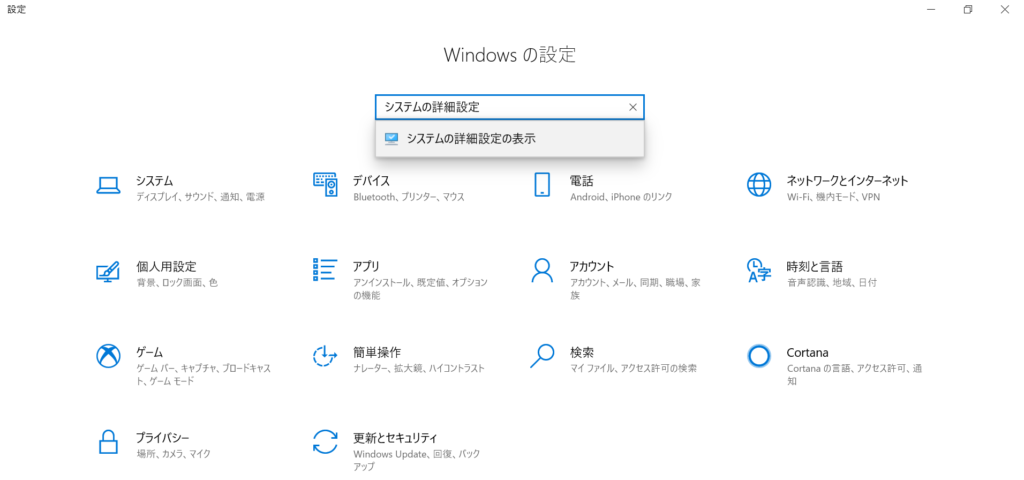
開いたら右下の環境変数をクリックします。
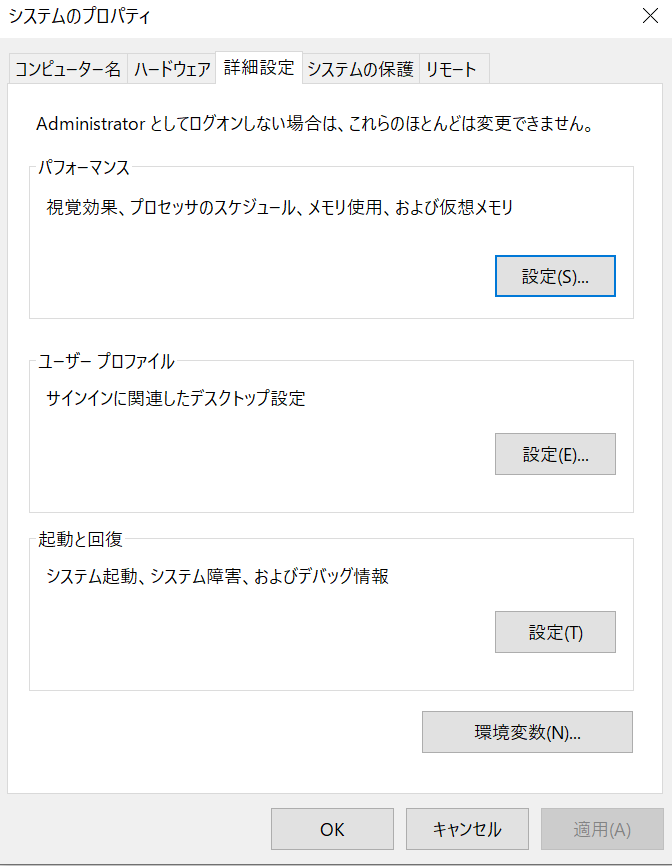
開くと同じものが上下に出てきますが、今回は上側のユーザーの環境変数の中にあるPathを編集します。Pathの項目をダブルクリックしてください。
すると、環境変数名の編集というウィンドが現れるので、右側の新規をクリックして、C:\MiFish\PMiFish\Tools (バックスラッシュは”円マーク”に変換)と記入後、OKをクリックします。
これで、パスの設定は終了です。
参考:https://www.atmarkit.co.jp/ait/articles/1805/11/news035.html
PMiFishのフォルダ構造
上記の作業が終わったPMiFishフォルダは以下のような構成になっていればOKです。
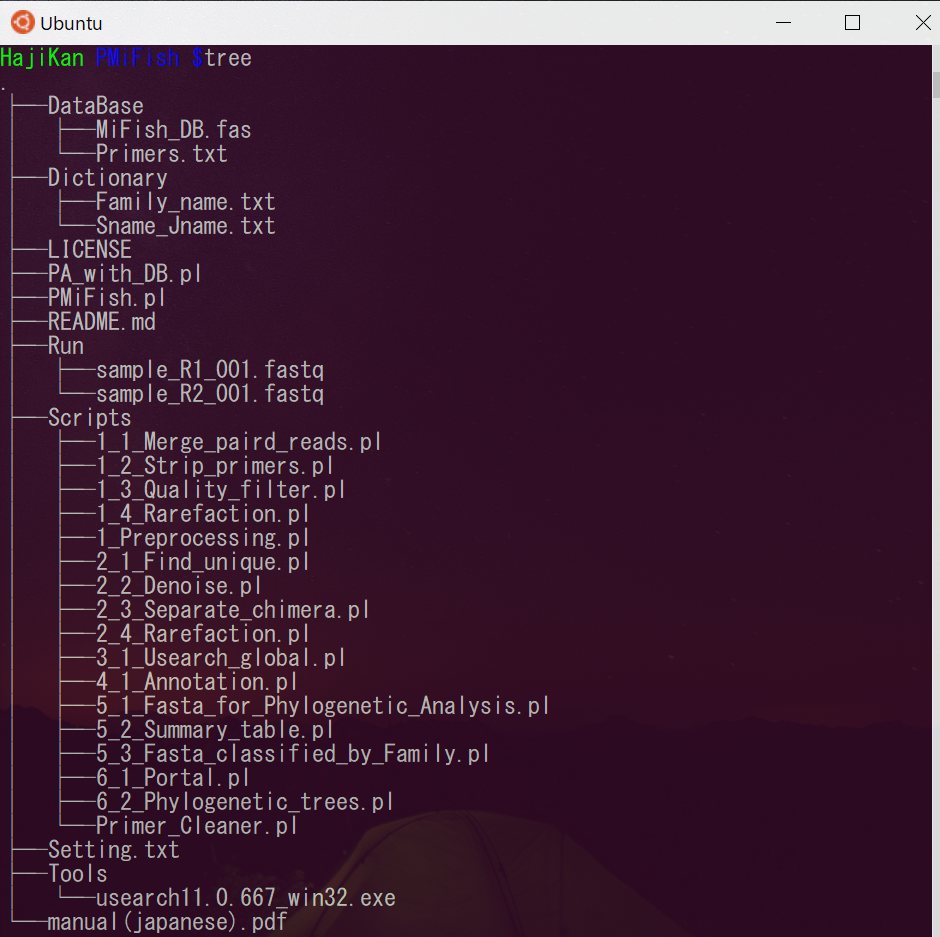
各フォルダの役割は以下の通りです。
- DataBase
– リファレンスデータ(Fasta形式)とprimer配列を記入したファイル - Dictionary
– 日本語、英語の種名辞書データ、および科名辞書 - Results
– 結果が出力されるフォルダ - Run
– 解析したいfastqファイルもしくはgzファイルを入れるフォルダ - Scripts
– ステップごとのスクリプトが入っているフォルダ
– ステップごとに解析 したい時に使用 - Tools
– Usearch および MEGAの設定ファイル(.mao)を入れておくフォルダ - PMiFish.pl
– 解析を走らせるスクリプト - PA_with_DB.pl
– PMiFish.pl の 解析後に実行することで、科レベルの系統樹を作成
– 結果は5-2 Summary_ table内に出力 さ れ る - Setting.txt
– 各種設定を記入するファイル
PMiFishを動かすための設定
PMiFishのパラメーター値の設定はSetting.txtにて行います。中身は以下のようになっています。
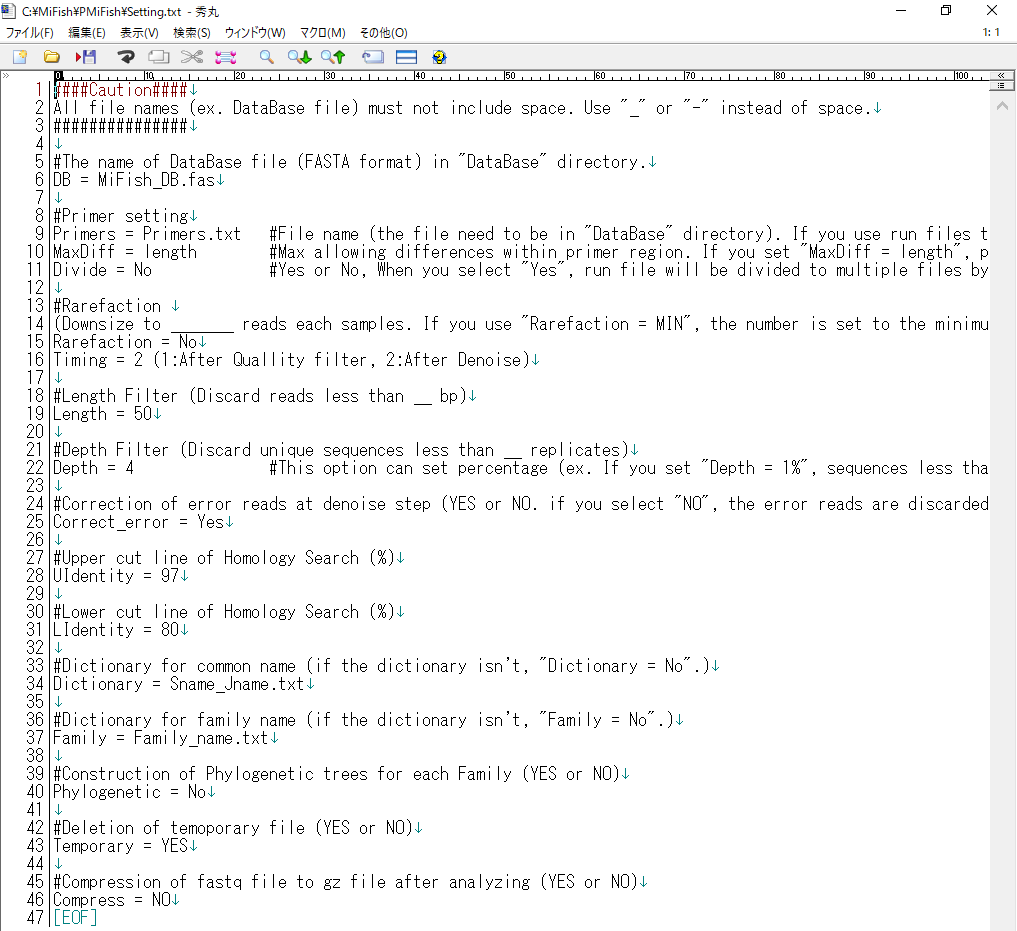
全体の編集ルールとして以下のことを守ってください。
- ファイル名にスペースは使用しない(アンダーバーで代用)
- #は説明文の意味なので消さない
- 半角英数のみ使用
Setting.txtの内容は以下の通りです。基本的にデフォルトの設定でいいと思います。
オレンジで示した部分は解析に使用される部分です。
- リファレンスファイル(データベースファイルの指定)
– DataBase フォルダ中にあるリファレンス ファイル(Fastaフォーマット)を指定する
DB = ファイル名
例: DB = MiFish_DB.fas - 使用したプライマー配列
– Databaseフォルダ中にあるプライマー配列が記入されたファイルを指定
– プライマー配列がすでに除去されているFastqファイルを解析する場合はNOを使用
Primers = ファイル名
例:Primers = Primes.txt or Primers = NO - プライマー領域中の最大ミスマッチ数の指定
– 1つのプライマーペアしか使用していないならlengthを指定するのがおすすめ
MaxDiff = 数値 or length - 複数のプライマーペアで増幅されたリードの場合、それを分けるかどうか
– Yesなら以降の解析は別々に処理
Divide = YES or NO - Rarefactionの設定
– リード数を希釈化する場合は数値もしくはMIN、しない場合はNO
– MINは最もリード数が少ないサンプルに合わせる
Rarefaction = 数値 or MIN or NO - Rarefactionの実行タイミングの設定
– Quality filterの後に実行する場合は 1
– Denoiseの後に実行する場合は 2
Timing = 1 or 2 - 解析に使用する配列の長さの設定
Length = 数値 - 同じ配列がいくつあれば解析に使用するかの設定
Depth = 数値 or 百分率
例1: 同じ配列が4以上あるものを使用 Depth = 4
例2: 各サンプルの全リード数に対して、指定した百分率から算出 Depth = 0.01% - Denoiseでエラーとされた配列を補正して以降の解析に使用するかの設定
Correct_error = YES or NO
例1: 補正して使用 Correct_error = YES
例2: すべて削除 Correct_error = NO - 相同性の設定
UIdentity = 数値
例1: 相同性が97%以上のものを採用する場合 UIdentity = 97 - 最低の相同性の設定(指定値以下は不明とする)
LIdentity = 数値
例1: 相同性が80%より低いものは不明とする LIdentity = 80 - 標準名の辞書を設定(辞書はDictionaryフォルダ内に配置)
Dictionary =ファイル名 or NO
例1: Dictionary = Sname_Jname.togodb.nonredundant.txt
例2: 使用しない Dictionary = NO - 科名辞書を設定(辞書はDictionaryフォルダに配置)
Family =ファイル名 or NO
例1: Family = Family_name_Fish.txt
例2: 使用しない Family = NO - 科ごとに系統樹を作成するかどうか(作成するためにはMEGAXが必要)
Phylogenetic = YES or NO - Preprocessingのファイルを解析後に削除するかどうかの設定
Temporary = YES or NO - Fastqファイルを圧縮するかどうかの設定
Compress = YES or NO
いくつかの変数についてのコメントです。
リファレンスデータベース①について
MiFish領域の配列はかなりのスピードで更新されています。データベースの拡充度合いによって結果が変わってくるので、NCBIから独自に配列を取得する場合はバージョン管理をしっかりとしましょう。
使用したプライマー②について
淡水だと基本的にMiFish-U-F/Rを単体で使用するか、検出しにくい種(ヤツメウナギやアユ)用にカスタムされたプライマーを併用、汽水域や海域だとアナハゼや板鰓類用のU2かEv2を併用する場合があると思います。各プライマーの長さや配列の違いによって解析結果が変わってくるので、使用したプライマーが選択されているかを都度確認しましょう。
同じ配列がいくつあれば解析に使用するかの設定⑧について
シングルトン、ダブルトン、トリプルトンは最低でも除きたいので、4以上の数値を設定することをお勧めします。また、ハイスループットシーケンサーで解読されるリードの総量は都度変化するので、固定値ではなく全体の中での割合や、近縁種とのリードの関係性も含めて結果を評価するのがいいと思います。
相同性の設定⑩について
相同性の値はMiFishの場合、可変領域の長さの平均が172bpなので1塩基の違いを許容する場合は(171/172)*100 = 99.41%、2塩基の違いを許容する場合は(170/172)*100 = 98.83%のように、自分の中で1種として何塩基違いを許容するかという観点のもとで決定していきます。
また、100%一致したものとそれ以外とで結果を出力してから、精査を加えていってもいいと思います。
科ごとに系統樹を作成するかどうか⑭について
MEGAXというソフトウェアをインストールして設定ファイルを出力させた後に解析をする必要がありますが、ここではその手順については掲載してません。
PMiFishを動かしてみる
ここまで来たら後はコマンド1つで実行できます。まずはSetting.txtは何も編集せずに動かしてみます。大文字小文字は区別して実行しましょう。
|
1 |
$ perl PMiFish.pl |
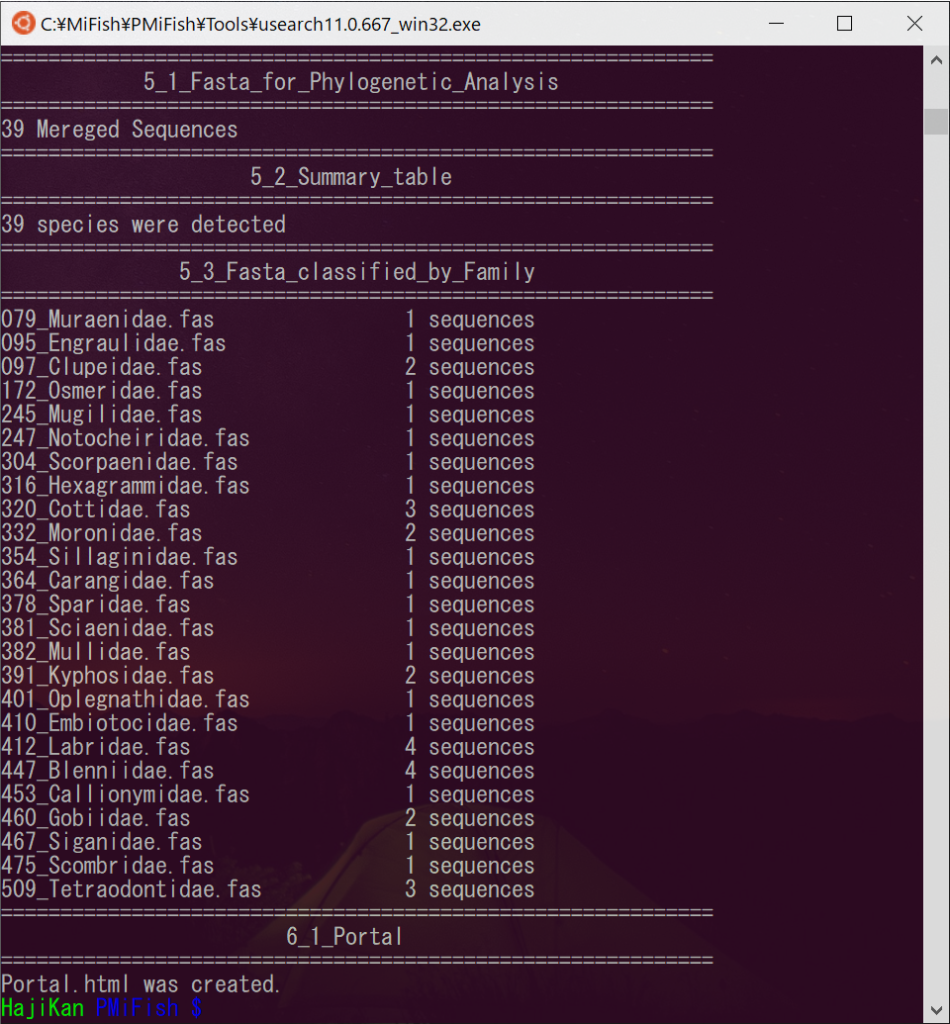
出力された結果を見てみる
Resultsフォルダには各処理段階での出力ファイルがまとめられます。
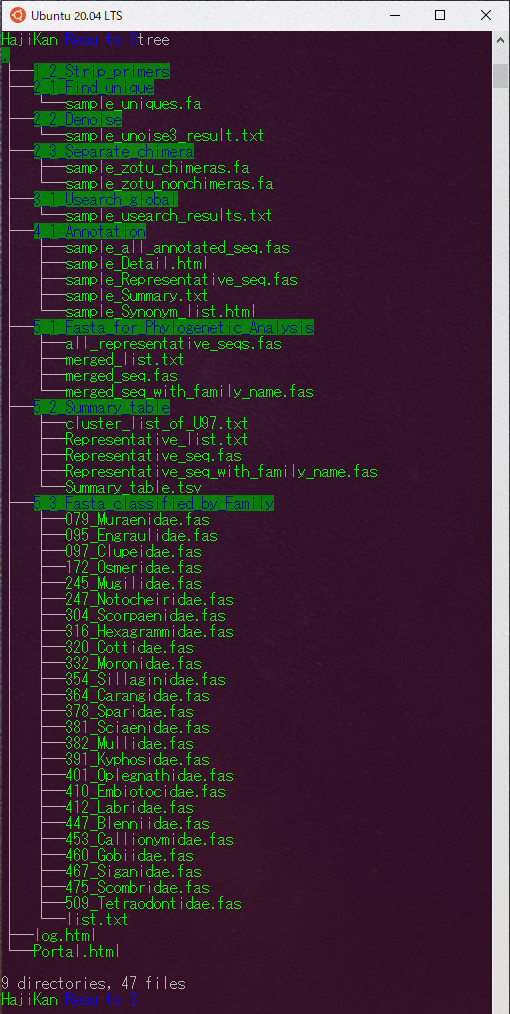
マニュアルの2,3ページにも各出力結果がどのようなものなのかが記載されています。
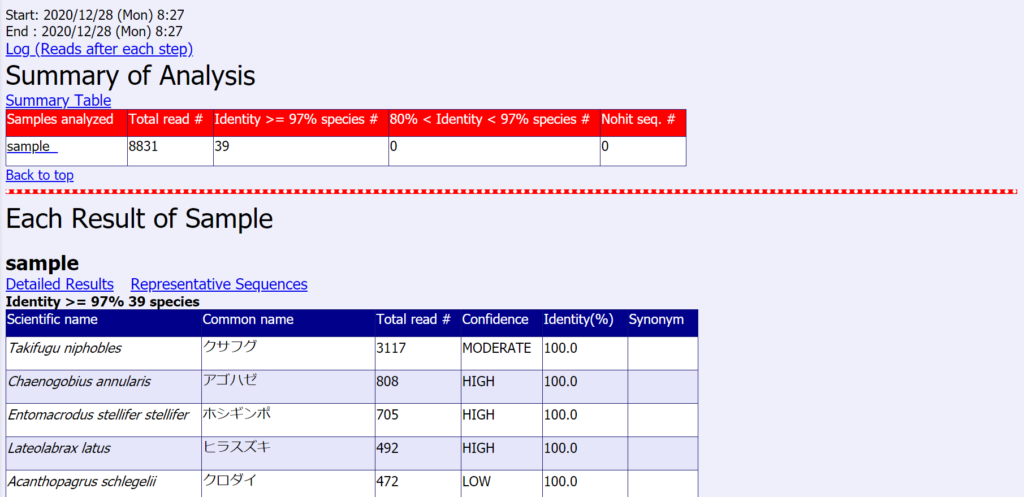
さっと結果を見るだけであれば、Portal.htmlを覗いてみるといいです。
Resultsフォルダの中で後のデータ解析や種の精査で特に必要なものは以下になります。一部抜粋して解説します。
- 2_3_Separate_chimera
- 4_1_Annotation
- 5_1_Fasta_for_Phylogenetic_Analysis
- 5_2_Summary_table
2_3_Separate_chimera
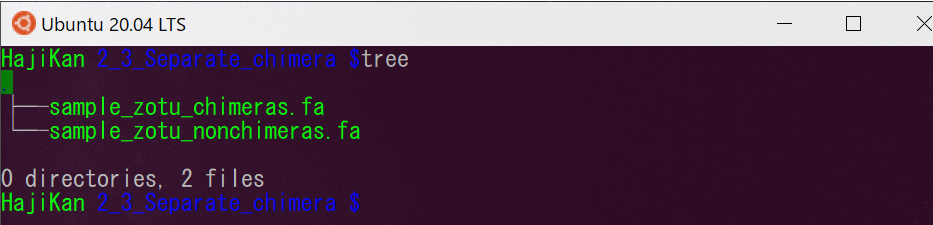
(サンプル名)_zotu_chimeras.faはキメラ配列が記載されているファイル
(サンプル名)_zotu_nonchimeras.faはキメラ配列を除去した配列が記載されているファイル
自前でのデータベースの作成はできないけど、最新のデータによる相同性検索がしたい!という場合には、NCBIのBlast検索を使用するといいと思います。その際には、ここの~nonchimeras.faを使用するといいと思います。
種の同定基準は環境省生物多様性センターの基準や精査方法に従うといいと思います。注意点として、時折誤同定かもしれないデータが混じっているので、系統樹などを見ながら種の精査を行いましょう。
Blast検索と出力結果の見方までを以下に記載します。
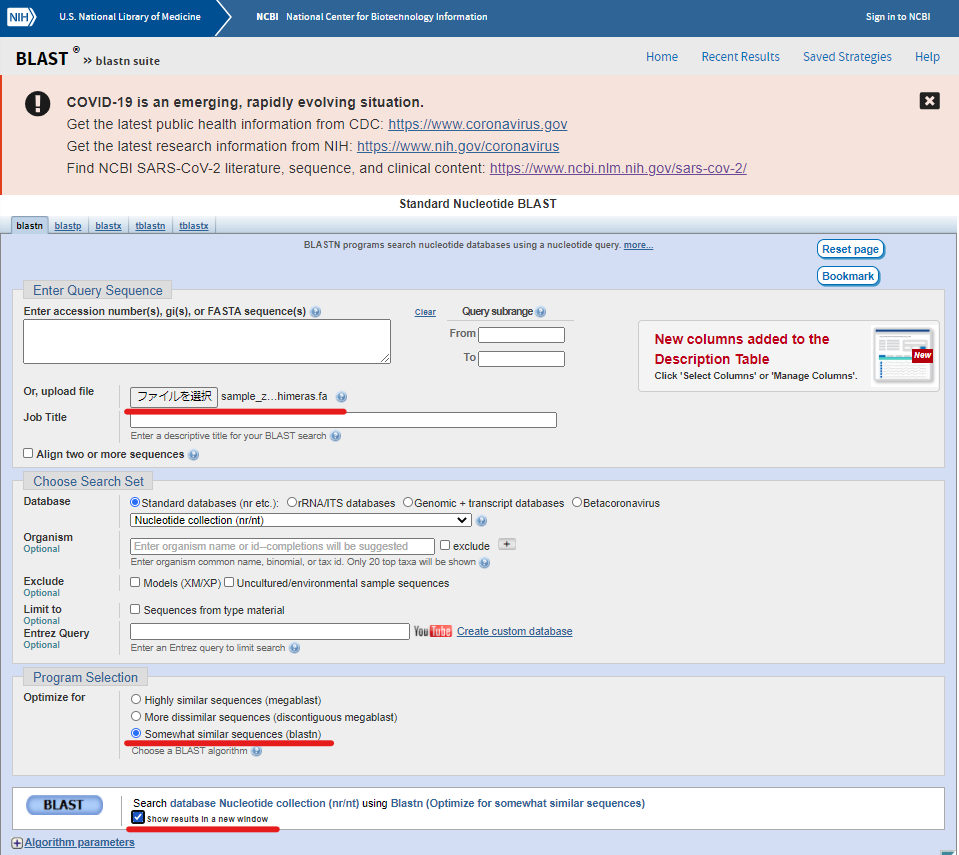
1. ここからBlastのページへ移動
2. 上段のファイルを選択より、~nonchimeras.faのファイルを選択
3. 下段のSomewhat similar sequences (blastn)を選択します。
megablastを使用してもいいですが、そんなにデータ量が大きくなったりするわけでもないので、環境DNAで使用する場合には、Somewhat similar sequences (blastn)を使用した相同性検索でいいと思います。
4.最下段のShow results in a new windowにチェックを入れることで、別タブに相同性検索の結果を出力してくれるようになります。
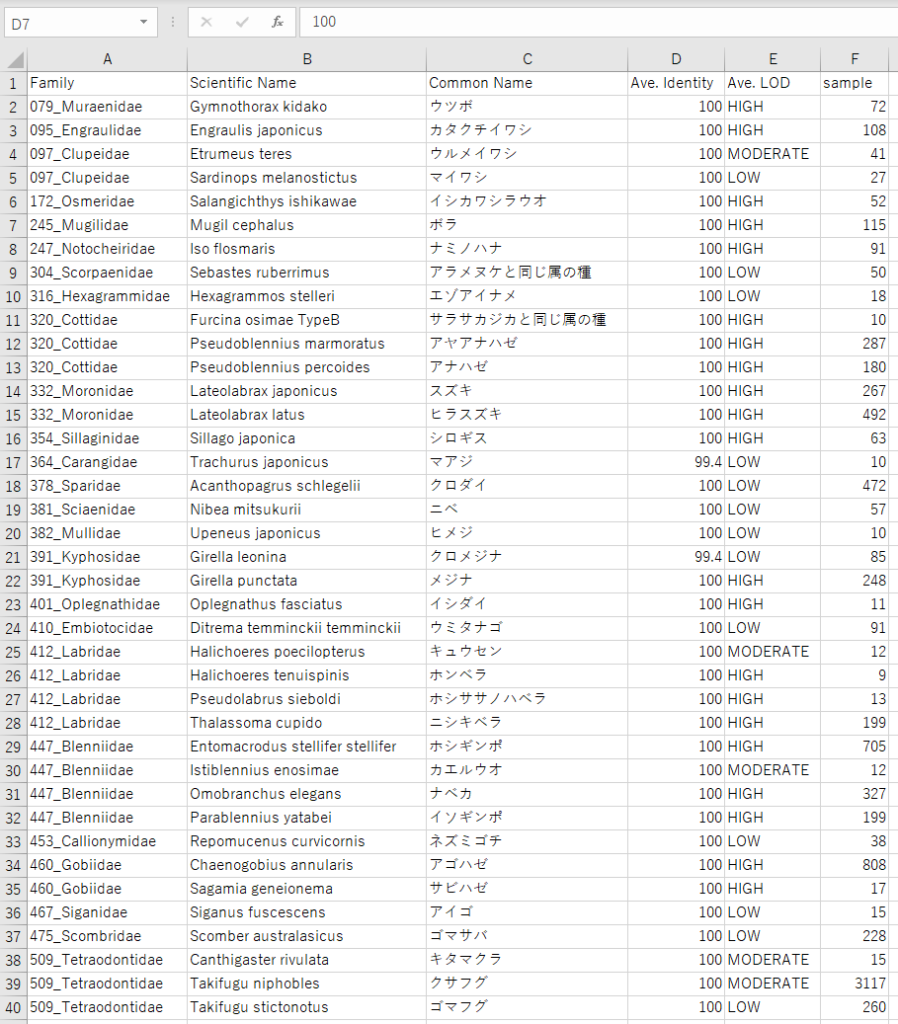
5. 以下のような結果が出力されます。
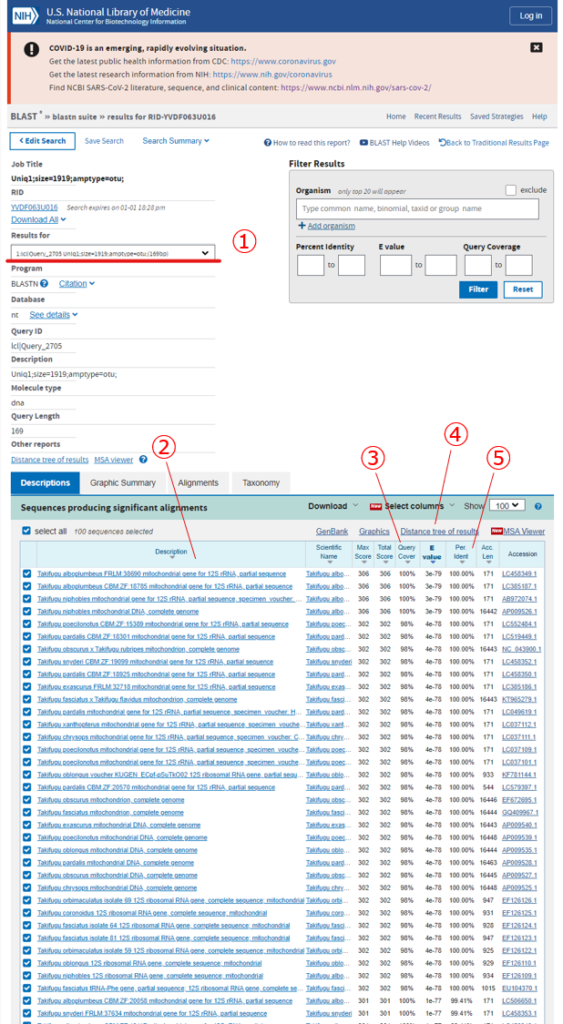
- ~nonchimeras.faにある配列名です
ここを変えることで~nonchimeras.fa内の各配列のBlast検索の結果が表示されます - 検索をかけた配列にヒットしたデータベースの配列の登録種名情報です
- 検索をかけた配列の全長とデータベースの配列が一致している領域の割合です
MiFishのメタバーコーデイングならここは100%であるものを優先して選ぶべきです - 検索をかけた配列とヒットしたデータベースの情報を用いて、系統樹を作成します
同じクレード内に別種が含まれてないか(配列を共有していないか)など確認したりします - 検索をかけた配列がデータベースの配列とどの程度一致しているか(相同性)を表しています
ですが、手作業で全サンプル分の精査を行うのは時間と労力の面でかなりパフォーマンスが悪いです。
分類群の特徴が分かる人(種を見ると怪しさが分かる人)が解析と種の精査を行うか、パイプラインのアップデートを自身で行うのがいいかもしれません。
4_1_Annotation
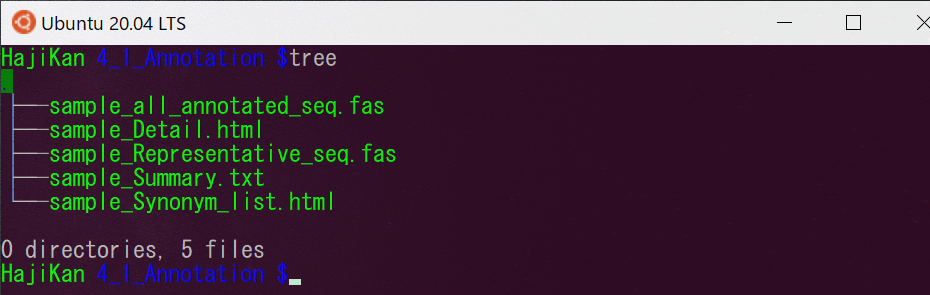
アノテーションとはデータに対して情報を付与することです。よってこのフォルダにはパイプラインの処理によって精査された配列に対して、データベースとマッチした種の名前やその確からしさなどの情報がまとめられています。
sample_Detail.htmlには下記のような検出された学名やリード数、信頼度や対象となる配列に対して、2番目に類似する種などの情報がhtml形式で記載されています。
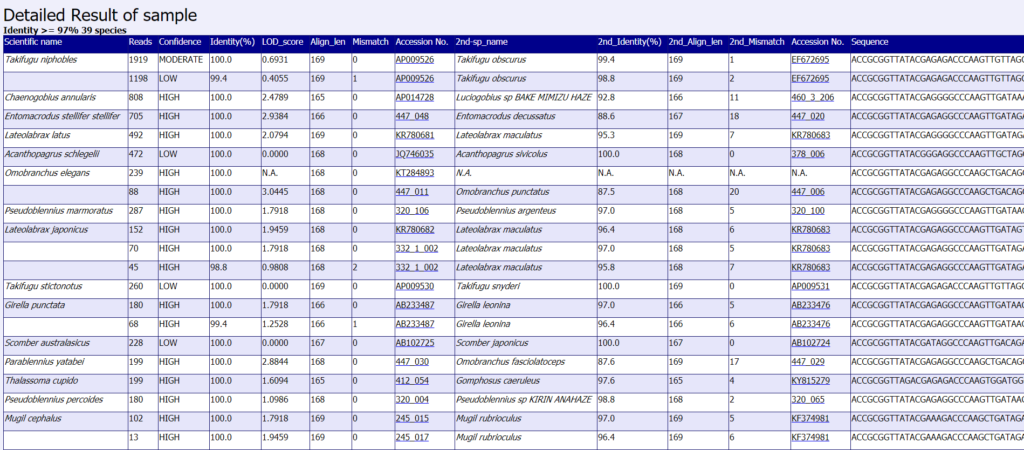
信頼度の指標として用いられているLow, Modelate, HighというのはwebのMiFishパイプラインでも出てきている指標です。種の精査の目安にします。
5_1_Fasta_for_Phylogenetic_Analysis
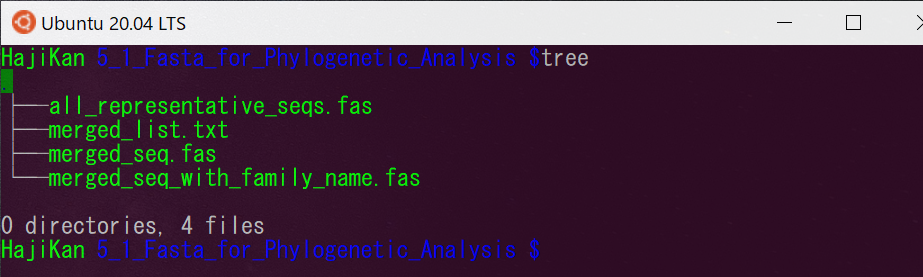
Phylogenetic analysisなので系統解析に使用するようなFastaファイルが入っています。
5_2_Summary_table
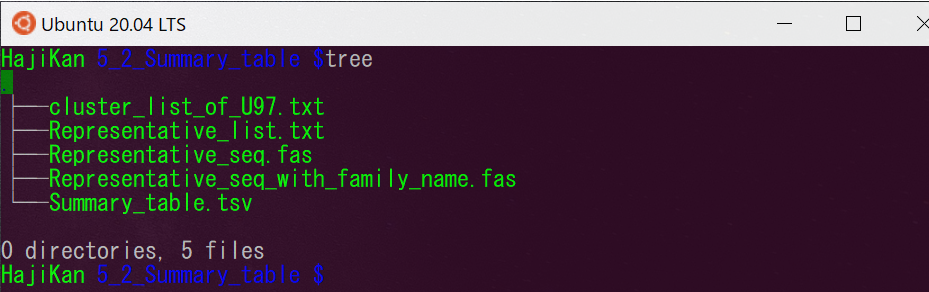
cluster_list_of_U97.txtはsetting.txtで設定した相同性の値(UIdentity)より下回った配列の情報がまとめられたファイルです。例に用いたデータにはそのような配列が無いみたいです。
Representative_list.txtとRepresentative_seq.fasはその種と同定した際に使用した配列とその情報がまとめられています。
Summary_table.tsvには分析したサンプルを横軸に、検出された種の情報が縦軸という形式でまとめられています。4_1_Annotationフォルダの情報を操作しやすいファイル形式にまとめた形になります。
エラーについて
Q. VCOMP140.DLLが見つからない
プログラムや起動に必要なパッケージが入っていないことから発生するエラーです。
見つからないと言われたvcmp140.dllはVisual Studio 2015 の Visual C++ 再頒布可能パッケージに含まれているそうです。
Usearchのサイトで案内されているリンクからダウンロードします。
32bitで提供されているフリーバージョンのusearchをダウンロードしたので、32bitバージョンのパッケージをダウンロードします。
1分くらいでインストールが終わります。エラーが出たタイミングに戻って再度実行してみてください。
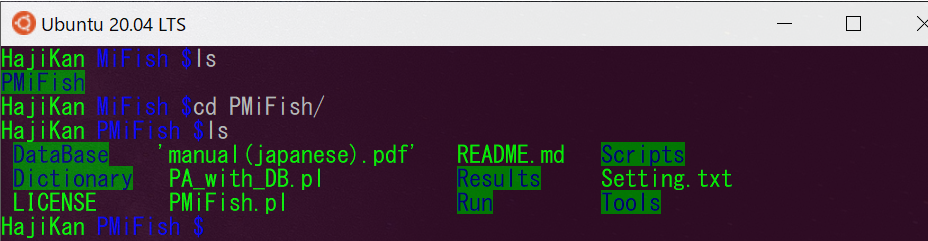
Q. perl PMiFish.plと打っても実行されない
このコマンドは自分がいるディレクトリにあるperlという言語で指示が書かれたPMiFish.plを実行するという意味です。
ということは、PMiFish.plというファイルがある階層で実行しないといけません。Ubuntu上で自分のいる場所(ディレクトリ)を一度確認してみてください。
|
1 2 |
# ディレクトリ内のファイルやフォルダを確認 $ ls |

|
1 2 3 4 5 |
# もしPMiFish.plが無ければ、cdコマンドで移動する # MiFishフォルダにいる場合、PMiFishとだけ表示されるので、PMiFishに移動する場合 $ cd PMiFish # ディレクトリ内のファイルやフォルダを確認 $ ls |
|
1 2 |
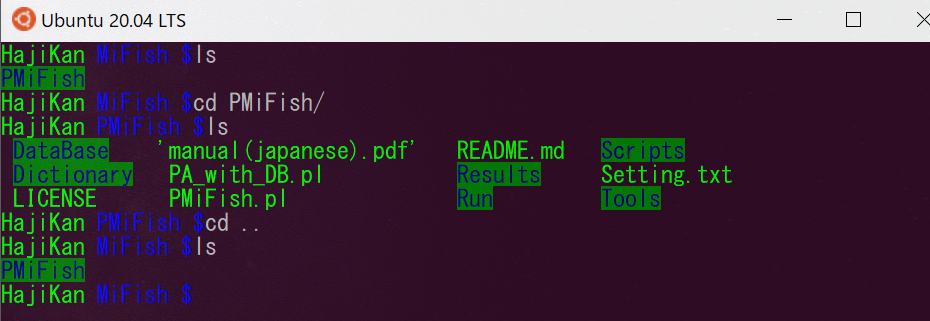
# 一つ前のフォルダに戻る場合の操作 $ cd .. |
Q. wgetでusearchがインストールされ始めない
会社や大学の場合、プロキシサーバーによる問題が考えられます。既にインターネットの接続が出来ている場合、.wgetrcにプロキシの設定(通信を許可してもらう設定)を書き込む必要があります。
参考URLに従って設定していただくか、ネットから直接ダウンロードしてくる方法(1)でトライしてみてください。
参考:https://takami-hiroki.hatenablog.com/entry/20101019/p1
Q usearchがうまく動いてなさそう
もし下記に類似するようなエラーが出たら、linuxの言語設定が出来ていないのかもしれません。
|
1 2 3 4 5 6 7 |
perl: warning: Setting locale failed. perl: warning: Please check that your locale settings: LANGUAGE = (unset), LC_ALL = (unset), LANG = "en_US.UTF-8" are supported and installed on your system. perl: warning: Falling back to the standard locale ("C"). |
この際、日本語化しとくといいかもしれません。私も同様のエラーに遭遇したので参考URLの内容に従って、WSLのUbuntu環境を日本語化していきます。
|
1 2 3 |
# パッケージ情報の更新 $ sudo apt update $ sudo apt upgrade |
|
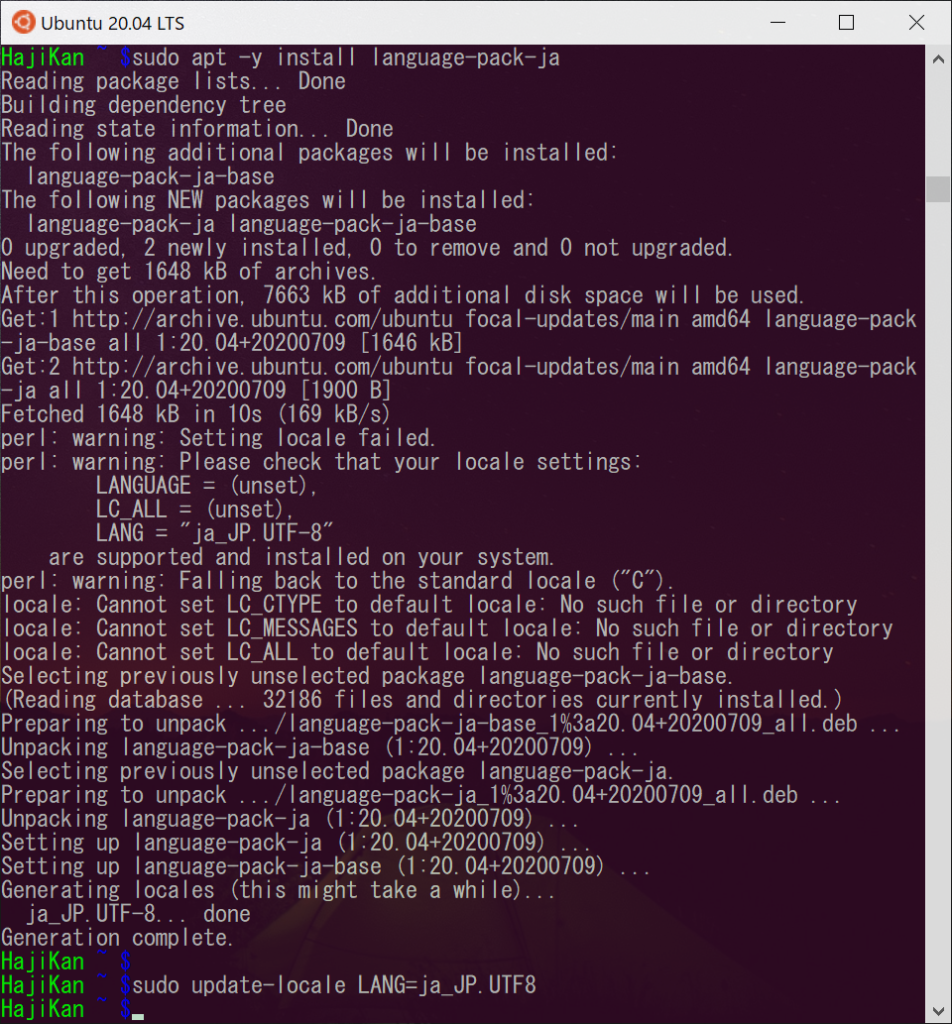
1 2 3 4 |
# 日本語言語パックのインストール $ sudo apt -y install language-pack-ja # ロケールを日本語に設定 $ sudo update-locale LANG=ja_JP.UTF8 |
|
1 2 |
# ここまできたら、Ubuntuを落としてから再起動 $ exit |
|
1 2 3 4 |
# タイムゾーンをJSTに設定 $ sudo dpkg-reconfigure tzdata # 日本語マニュアルのインストール $ sudo apt -y install manpages-ja manpages-ja-dev |
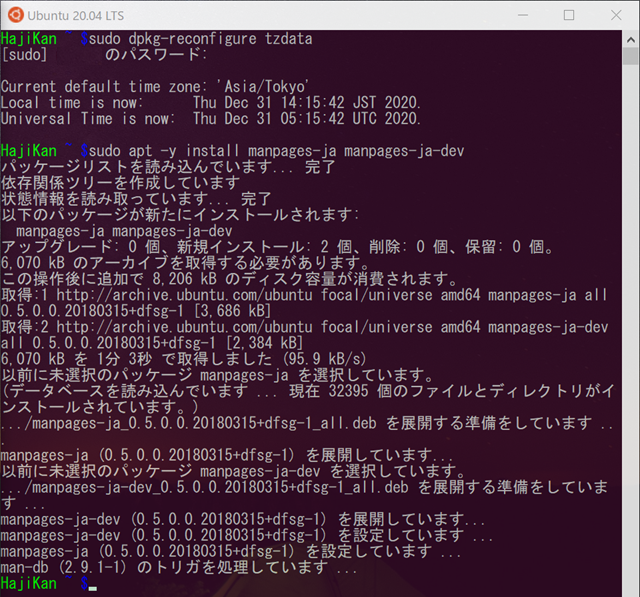
これで日本語設定になりました。
参考:https://www.atmarkit.co.jp/ait/articles/1806/28/news043.html
ちなみに、やっぱり英語設定の方がいい場合は下記のようにします。日本語設定にしたのちに、英語設定に戻しました。
参考URLに従って作業をしていきます。
|
1 2 |
# 現在の設定を確認する $ echo $LANG |

|
1 2 |
# 利用可能なロケール名の一覧を取得 $ locale -a |

言語の設定を変更するには update-localeコマンドを使用する
|
1 2 3 4 |
# 管理者権限をつけて実行 $ sudo update-locale LANG=en_US.utf8 # 変更後の内容を確認 $ cat /etc/default/locale |
|
1 2 |
# いったんUbuntuを落としてから、再起動 $ exit |
|
1 2 |
# ubuntuを再起動後、確認 $ echo $LANG |

以上、言語の設定でした。
参考: https://www.atmarkit.co.jp/ait/articles/1610/14/news033.html
Reference
- Komai et al. 2018
Development of a new set of PCR primers for eDNA metabarcoding decapod crustaceans - Miya et al. 2020
MiFish metabarcoding: a high-throughput approach for simultaneous detection of multiple fish species from environmental DNA and other samples - Oka et al.2020
Environmental DNA metabarcoding for biodiversity monitoring of a highly-diverse tropical fish community in a coral-reef lagoon: Estimation of species richness and detection of habitat segregation - usearch v11
- ActivePerl
- ubuntuで(できるだけ)全部のlocaleを追加する -Qiita
- Windows10でPath環境変数を設定/編集する
免責事項
十分注意は払っていますが、本記事の情報・内容について保証されるものではありません。本記事の利用や閲覧によって生じたいかなる損害について責任を負いません。また、本記事の情報は予告なく変更される場合がありますので、ご理解くださいますようお願いします。