

Mitohelper:魚類の環境DNA研究のためのミトコンドリア参照配列分析ツール
この記事でやりたい事と目的
Mitohelperでどんなことができるのか見ていきます。
論文要約
魚類の環境DNAメタバーコーディングは非侵襲的で費用対効果の高い方法です。
ですが、多くの魚種はまだ参照データベースとしてシーケンスされておらず、魚類を対象とした環境DNAのリソースの開発は十分ではありません。
既存のMitoFishデータベースで利用可能な魚類のミトコンドリア配列に注釈(アノテーション)を付けて整形するPythonベースのコマンドラインツールであるMitohelperを開発しました。
Mitohelperは遺伝子名と追加の分類学的分類を追加することにより、MitoFishのアノテーションを改善します。
Mitohelperのgetrecord関数は、これらの参照データセットを使用して、ユーザーが用意したの魚類の分類名のリストに対して、MitoFishで利用可能なミトコンドリア遺伝子(COI, 12s rRNA, その他) を検索します。
Mitohelperのgetalignment関数は、重複するシーケンス領域の評価と視覚化のために、ミトコンドリア遺伝子配列 (大半はミトコンドリアDNAの一部の領域) をユーザーが指定した完全長参照配列にアラインメントします。
各ツールにおける分子同定を容易にするために、Mitohelperの12S rRNAデータセットをQIIME2の共通フォーマットでSILVAの16SrRNAおよび18SrRNAデータセットと組み合わせました。
Mitohelperは環境DNAデータのリソース幅、機能、および分類学的分類の精度を向上させます。
Mitohelperとその参照データセットはほぼ毎月更新されます。
https://github.com/aomlomics/mitohelper
Mitohelper
Mitohelperは魚類の環境DNA研究用のpythonベースのミトコンドア参照配列分析ツールです。以下の条件に当てはまる人には役に立つかと思います。
- 特定の魚種や分類群のミトコンドリアDNA参照配列が存在するかを調べる
(getrecordコマンドを使用) - ミトコンドリア遺伝子のどの領域が配列決定されているかを長鎖の配列に対してアライメントすることで見つける
(getalignmentコマンドを使用) - RESCRIPtを使用することで、QIIMEで使用できる遺伝子参照データベースを作成し、下流の解析に使用できる

Mitohelperを取得して使えるようにする
流れ
- 製作者のGitHubからzipファイルまたはgit cloneでファイル群を入手
- 依存ツールを入手
- Blast+ (local環境でblastを使用するため)
- python 3.9.1(本記事はpythonが使用できる前提)
- 要求されるpythonモジュールを入手
- click (v7.1.2)
- matplotlib (v3.3.0): グラフを作成するのに必要
- pandas (v0.25.3): データ整形をするのに必要
- seaborn (v0.10.1): グラフを作成するのに必要
以下の内容は下記のページでUbuntu-LTS 20.04をダウンロードしていることを前提として進めます。

1. Mithohelperのファイルを入手
作成者のgithubのページ(https://github.com/aomlomics/mitohelper)にいきます。
zipファイルをダウンロードする場合は、右上のCodeタブ内のDownload ZIPをクリックしてダウンロードします。ダウンロードしたら自分の作業ディレクトリに解凍して配置します。
私はcドライブ上にMitohelperというフォルダを作成してそこにダウンロードしたファイルを入れました。
git cloneを使用する場合は、Codeタブ内のHTTPSタブに記載されているURLをコピーして、下記コマンドを実行します。少し時間がかかります。
|
1 |
$ git clone https://github.com/aomlomics/mitohelper.git |
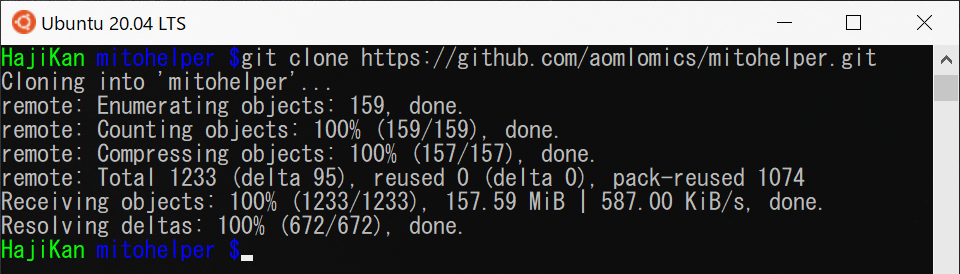
作成したmitohelperフォルダを起点としたフォルダ構造はこんな感じです。
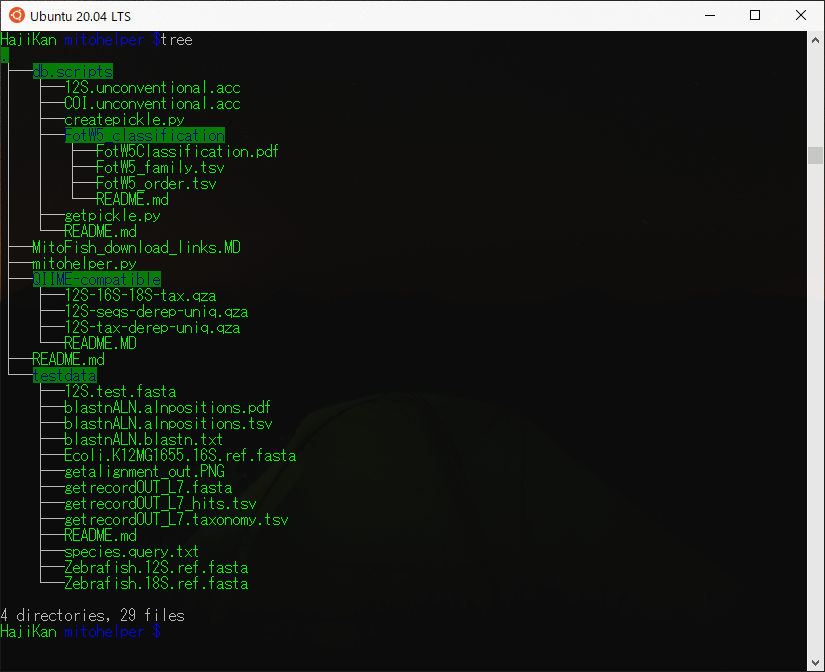
これでMitohelperの取得は終了です。
gitについてはここら辺が分かりやすいと思います。
2. 依存ツールを入手
BLAST+のインストール
Windows Subsystem for Linux (WSL)環境で、BLAST+をインストールします。
まず、Ubuntuを起動してパッケージリストとパッケージの更新をしておきます。
|
1 2 3 4 |
# パッケージリストの更新 $ sudo apt update -y # パッケージを更新 $ sudo apt upgrade -y |
パッケージするとを更新したら次にBLAST+をダウンロードします。
|
1 |
$ sudo apt install ncbi-blast+ -y |
ちゃんとBLASTが使用できるようになっているかhelpコマンドで確認してみます。
|
1 |
$ blastn --help |
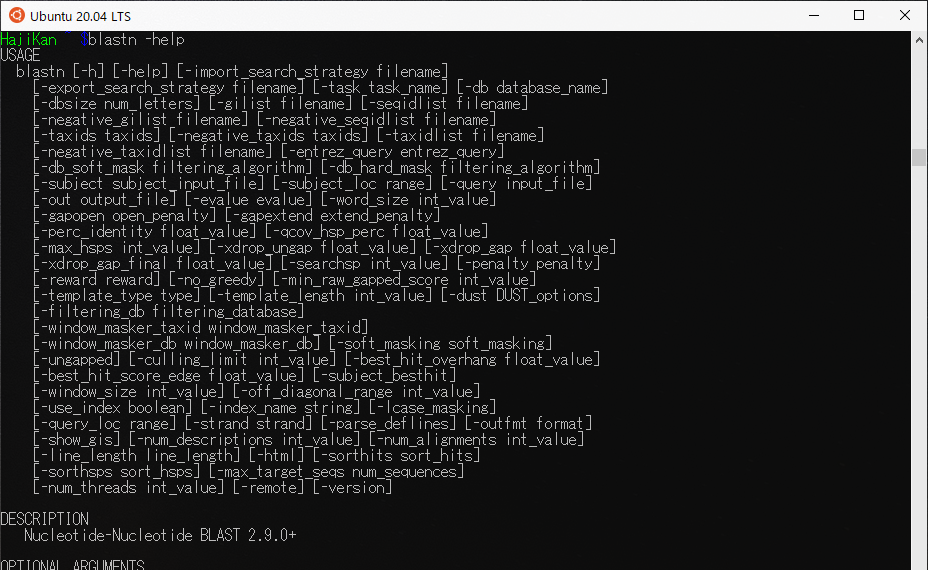
このようにコマンドがずらっと表示されたら無事BLASTがインストールされています。
Pythonのバージョンについて
論文はpython 3.6.10で動作させているようですが、私は3.8.5で動かしました。
python環境構築の細かいことについてはここでは記載しません。バージョンが3.6.10以上であれば問題ないと思います。
要求されるpythonモジュール
Mitohelperは以下のpythonモジュールを使用します。
- click (v7.1.2)
- matplotlib (v3.3.0): グラフを作成するのに必要
- pandas (v0.25.3): データ整形をするのに必要
- seaborn (v0.10.1): グラフを作成するのに必要
私はpipを使ってパッケージ管理をしているので、pipを使って一気にインストールしておきます。
|
1 |
$ pip3 install click matplotlib pandas seaborn |
これで必要なモジュールはそろったかと思います。
Mitohelperを使ってみる
”mitohelper.py”があるディレクトリ上で以下のコマンドを実行します。
|
1 |
$ python3 mitohelper.py --help |
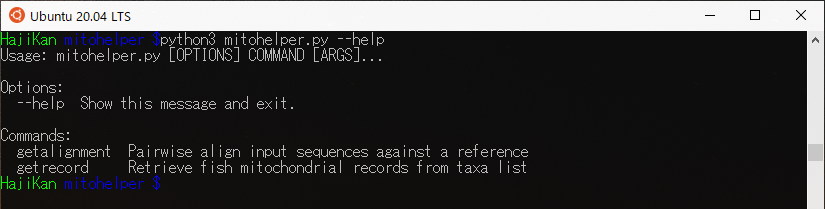
指定した魚種のレコードを取得するgetrecordコマンド
Usage exampleのgetrecordコマンドを例にしたがって動かしてみます。
getrecordコマンドはユーザーが指定する魚類の分類リストからシーケンスレコードをfasta形式もしくはQIIME形式で出力します。
helpコマンドでどのような機能があるのか見てみましょう。
|
1 |
$ python3 mitohelper.py getrecord --help |
getrecordの機能
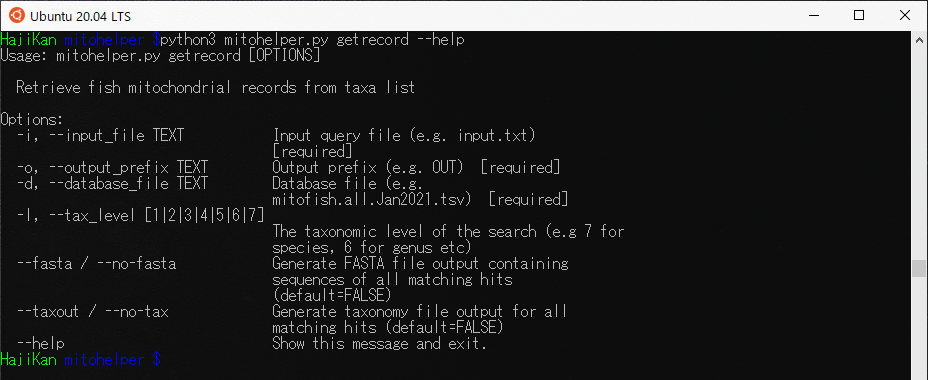
- -i : レコードが欲しい魚種名が1列に記載されているテキストファイルを指定[必ず必要]。
ex) -i testdata/species.query.txt - -o : 出力するファイルの名前を指定[必ず必要]
ex) -o testdata/getrecordOUT - -d : データベースファイルの名前を指定[必ず必要]
ex) -d mitofish.all.Feb2021.tsv - -l : 検索する分類学的レベルを指定[必ず必要]
7: species, 6: geneus, 5:family , 4:order , 3:phylum, 2:?, 1:?,
ex) -l 7 - –fasta : ヒットしたすべて配列をfasta形式で出力(デフォルトはFalse)
ex) –fasta - –taxout : ヒットしたすべての分類群が記載されたファイルをtsv形式で出力
ex) –taxout - –help : ヘルプメッセージを表示
指定した魚種のレコードをgetrecordで取得してみる
githubの例は’mitofish.all.Jan2021.tsv’のデータベースを指定していますが、動作させるタイミングによってデータベースの種類が異なる点に注意です。
この記事を書いたのは2021年2月です。データファイルなどがまとめられているこのページ(http://doi.org/10.5281/zenodo.4533286)から、’mitofish.all.Feb2021.tsv’をダウンロード(※1.2GBくらい)してmitohelper.pyと同じフォルダに配置します。
コマンドを動かす前に同一のファイル名のものが出力されるので、getrecord~と名前のついているファイルを削除しておきましょう。
|
1 |
$ rm testdata/getrecord* |
getrecordで要求する魚種名をspecies.query.txtに記載します。元から以下の5種は多分ですがわざと大文字や小文字を交えて記載されています。
- Abraliopsis pfefferi
- Ahliesaurus berryi
- Alepisaurus FEROX
- anotopterus pharao
- AHLIESAURUS BERRYI
また、そのままではなんか味気ないので、ドンコ(Odontobutis obscura)をリストに加えてgetrecordを実行してみます。
|
1 |
$ python3 mitohelper.py getrecord -i testdata/species.query.txt -o testdata/getrecordOUT -d mitofish.all.Feb2021.tsv -l 7 --fasta --taxout |
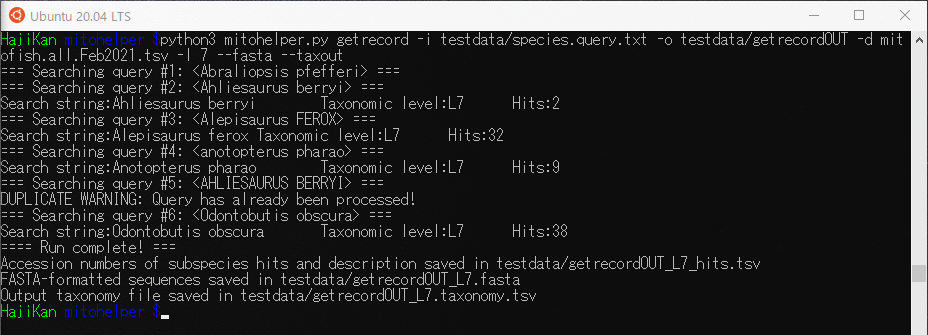
実行すると以下のファイルが生成されます。
- getrecordOUT_L7_hit.tsv
- getrecordOUT_L7.taxonomy.tsv
- getrecordOUT_L7.fasta
getrecordOUT_L7_hit.tsv
以下のような構成になっています。
- クエリー
- NCBIのアクセッションナンバー
- GenBankレコードで定義されている遺伝子名
- NCBIのtaxid
- superkingdom
- 門
- 綱
- 目
- 科
- 属
- 種
- 配列
- OrderID (“Fishes of the World, 5th edition” (Nelson et al. 2016))
- FamilyID(“Fishes of the World, 5th edition” (Nelson et al. 2016))
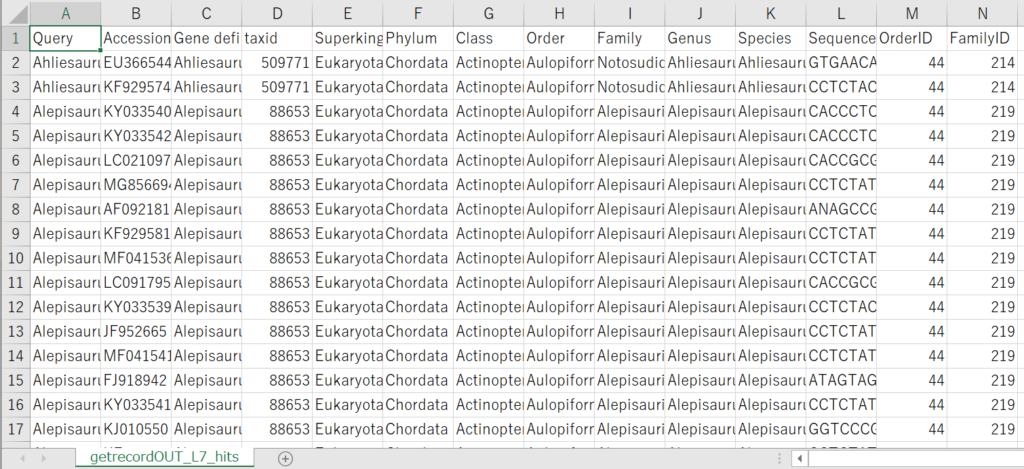
Queryの列は指定した種名、Accessionの列にはその種に対応するNCBIのアクセッションナンバーが配置されています。
getrecordOUT_L7.fasta
fastaファイルは処理が甘いのか完全なfasta形式になってなかったので、使用するなら処理を書き換える必要がありそうです。
以下のような感じでした。
>EU366544
getrecordOUT_L7.fasta
GTGAACATGAGGTGGGCTCAGACGATAAAGCCTAGGAGGCCGATTGCTATCATAGCTCAGACCATGCCCATGTAGCCAAAGGGTTCTTTTTTCCCTGAATAGTAG…ACGAAAGCGTGGGCAGTAACGATAACATTGTAAATCTGGTCGTCTCCTAAAAGGGCTCCGGGTTGGCTTAGCTCAGCTCGGATGAGAAGGCT>KF929574
CCTCTACCTACTATTTGGTGCCTGGGCCGGGATGGTGGGTACAGCCCTAAGCCTTCTCA….
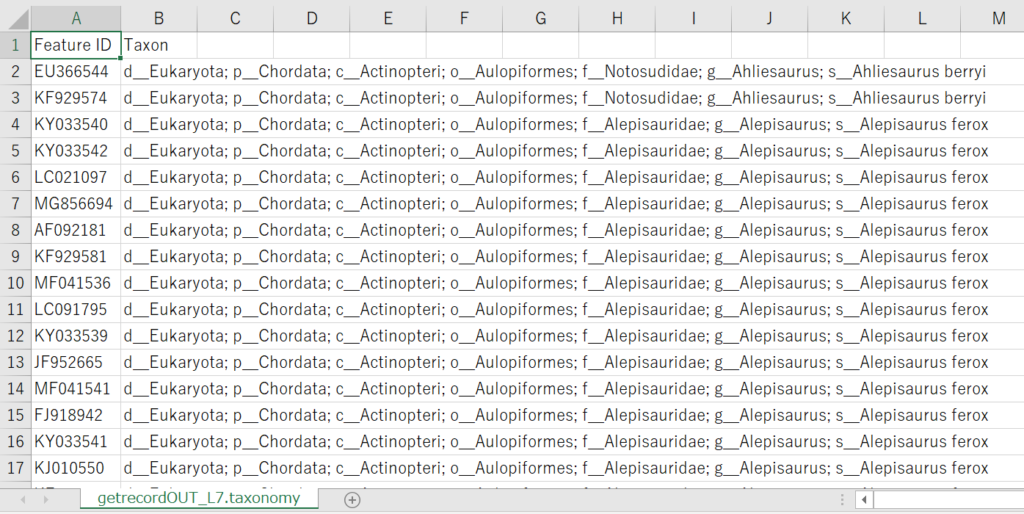
getrecordOUT_L7.taxonomy.tsv
getrecordOUT_L7.taxonomy.tsvはアクセッションナンバーとそれに対応する登録種名がQIIME形式のデータベースに準拠した表記法になっていました。
次は階級を指定するオプションを5にしてみます。
|
1 |
$ python3 mitohelper.py getrecord -i testdata/species.query.txt -o testdata/getrecordOUT -d mitofish.all.Feb2021.tsv -l 5 --fasta --taxout |
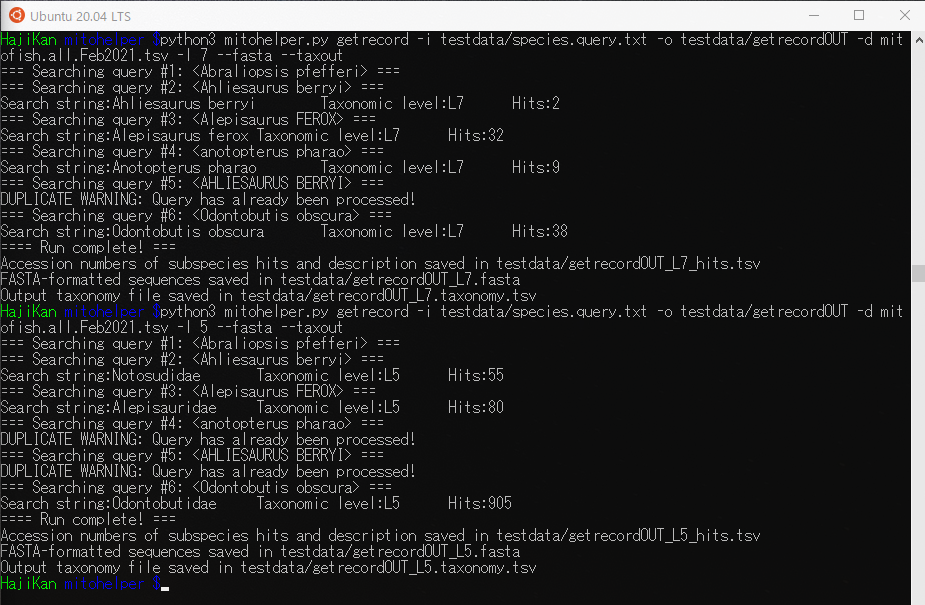
7を指定した時と比べてhit数が大きくなりました。
L5のHitファイルを見てましょう(※10列分のみ表示)。
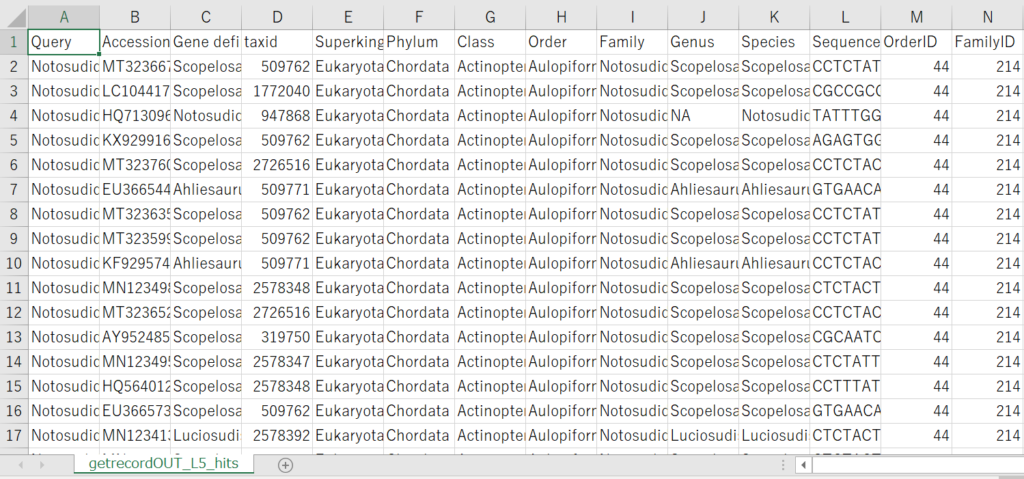
L7の時と比べて、Queryの列には、指定した種が属する科が、Accessionの列には科に属する種のNCBI アクセッションナンバーが記載されています。
なので、取得したい配列情報が対象となる種以外に、同属もしくは同じ科の近縁種まで欲しいという場合は、分類群のレベルを指定する-lの値を小さくしてあげればいいということのようです。
getalignment
Usage exampleのgetalignmentコマンドを例にしたがって動かしてみます。
getalignmentコマンドはユーザーが指定したfasta形式の参照ファイルからlocal blastの結果を使用して、アライメント位置をグラフとして出力します。
helpコマンドでどのような機能があるのか見てみましょう。
|
1 |
$ python3 mitohelper.py getalignment --help |
getalignmentの機能
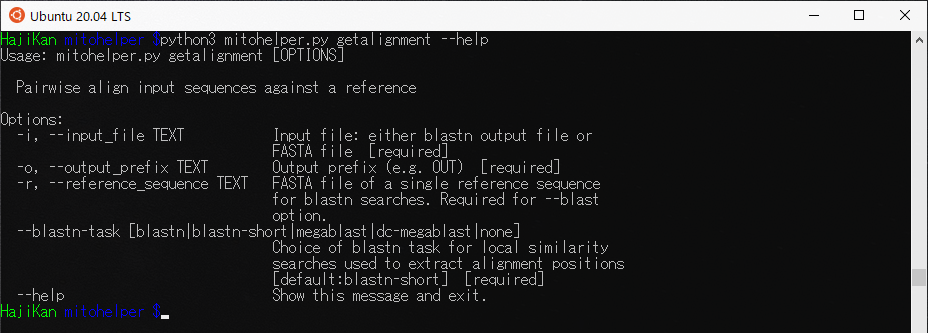
- -i : blastnの出力ファイルかfastaファイルのいずれかを指定[必要]
ex) -i testdta/12S.test.fasta - -o : 出力ファイル名を記載[必要]
ex) -o testdta/blastnALN - -r :blastnで使用される単一の配列が記載されたfastaファイルを指定
ex) -r testdta/Zevrafish.12S.ref.fasta - –blastn-task : blastnの検索法を指定[必要]
ex) –blastn-task blastn
核酸の相同性検索を行うblastnにも、blantn, blastn-short, megablast, dc-megablasstなどいくつかの最適化オプションがあります。
一般的な使用はblastnを使っておけばいいと思います。詳しくはNCBIのマニュアルを見てみてください。
getalignmentでアライメント結果を取得・視覚化してみる
入力に指定する12S.test.fastaはこのような形式です。一部省略してあります。
>AB938103
12S.test.fasta
CACCGCGGTTATACGAGTAACTCACATTAATACACCCCGGCGTAAAGAGTGATTAAAGAATGACCTTTAACTACTAA…
>AB006953
GATAACATCCCTATATGGTTTAGTACATAATATGCATAATATTACATTAATGCATTAGTACATATATGTATTAT..
>AB015962
GCTAGTGTAGCTTAATTTAAAGCATGGCACTGAAGATGCTAAGATGAGAAATAATTTTTTCCGCGGGCATA…
>AB016274
GTTATCGTAGCTTACTTCTAAAGCCTAGCCCTGAAAATGCTAAGATGGGCCCTAACGGCTCCGCCAACAAAA…
>AB018224
CATAAAGGTTTGGTCCTGGCTTTACTATCAACTCTAACCTGATTTACACATGCAAGTCTCCGCACTCC…
>AB018225
CATAAAGGTTTGGTCCTAGCTTTACTATCAACTCTAACCTGATTTACACATGCAAGTCTCCGCACTCCTGT…
>AB018226
CACAAAGGTTTGGTCCTAGCTTTACTATCAACTCTAACCTAATTTACACATGCAAGTCTCCGCATTCCTGT…
>AB018227
CACAAAGGTTTGGTCCTAGCTTTACTATCAACTCTAACCTAATTTACACATGCAAGTCTCCGCACTC…
>AB018228
CACAAAGGTTTGGTCCTAGCTTTACTATCAACTCTAACCTAATTTACACATGCAAGTCTCCGCACTCCT…
>AB018229
CACAAAGGTTTGGTCCTAGCTTTACTATCAACTCTAACCTAATTTACACATGCAAGTCTCCGCACTCCTG…
>AB018230
CACAAAGGTTTGGTCCTAGCTTTACTATCAACTCTGACCTAATTTACACATGCAAGTCTCCGCACTCCTGT…
参照配列となるゼブラフィッシュの12s領域の配列はこのような感じです。省略しましたが952bpあります。
>NC_002333.2:1020-1971 Danio rerio mitochondrion, complete genome
Zebrafish.12S.fasta
CAAAGGCATGGTCCCGACCTTTTGATCAGCTTTTACCTAATTTACACATGCAAGTCTCCGCACCCCTGT…
コマンドを動かす前に同一のファイル名のものが出力されるので、blastnALN~と名前のついているファイルを削除しておきましょう。
|
1 |
$ rm testdata/blastnALN* |
getalignmentコマンドを動かしてみます。
|
1 |
$ python3 mitohelper.py getalignment -i testdata/12S.test.fasta -o testdata/blastnALN -r testdata/Zebrafish.12S.ref.fasta --blastn-task blastn |
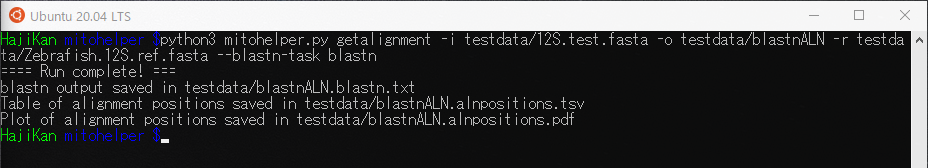
実行すると以下のファイルが出力されます。
- blastnALN.alnpositions.pdf
- blastnALN.alnpositions.tsv
- blastnALN.blastn.txt
blastnALN.blastn.txt
blastnALN.blastn.txtはBlast検索の出力結果ファイルです。
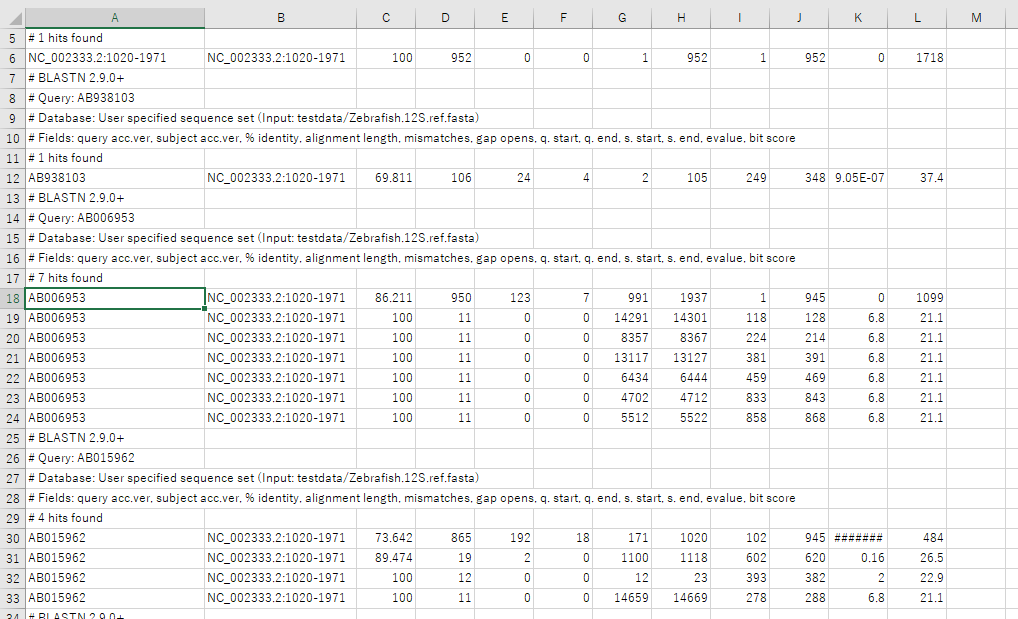
12S.test.fastaの11本の配列に対して、配列付与される情報は以下の12要素です。
| 要素名 | 意味 |
| query acc.ver | クエリのアクセッションナンバー |
| subject acc.ver | -rに指定した配列のアクセッションナンバー |
| % identity | 相同性 |
| alignment length | アライメント部の配列長 |
| mismatches | アライメントのミスマッチ数 |
| gap opens | ギャップの開始位置の数 |
| q.start | クエリ中のアライメントの開始位置 |
| q.end | クエリ中のアライメントの終了位置 |
| s.start | 検索対象の配列のアライメントの開始位置 |
| s.end | 検索対象の配列のアライメントの終了位置 |
| evalue | E-value |
| bit score | bit socre |
webでblast検索した時に得られるデータとほとんど変わりませんね。
blastnALN.alnpositions.tsv
blastnALN.alnpositions.tsvはblastnALN.alnpositions.pdfのグラフを書くためのデータファイルなのだろうと思います。
各入力配列に対するアライメント開始位置、終了位置、bit scoreが記載されています。
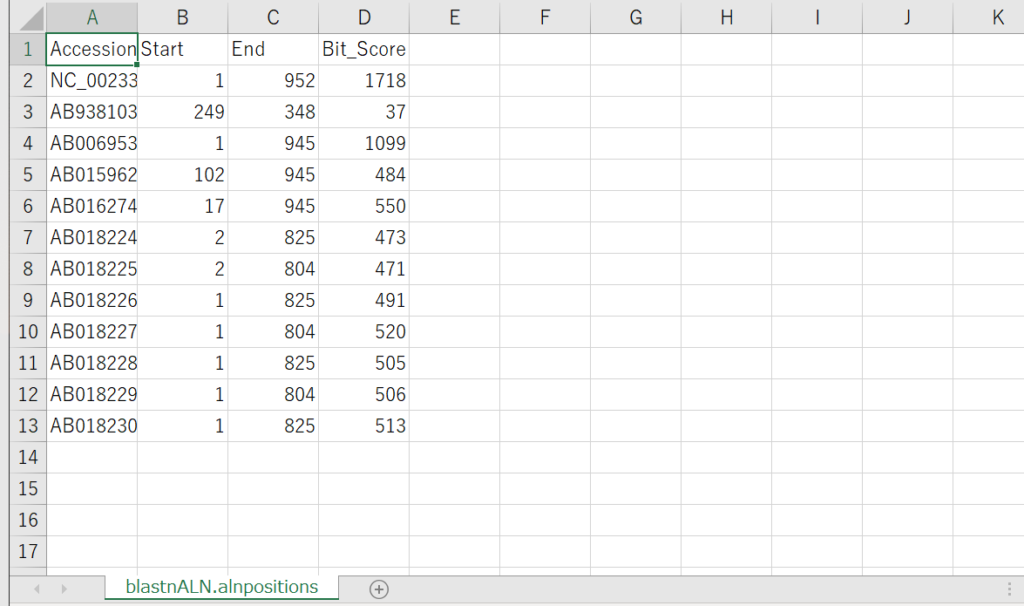
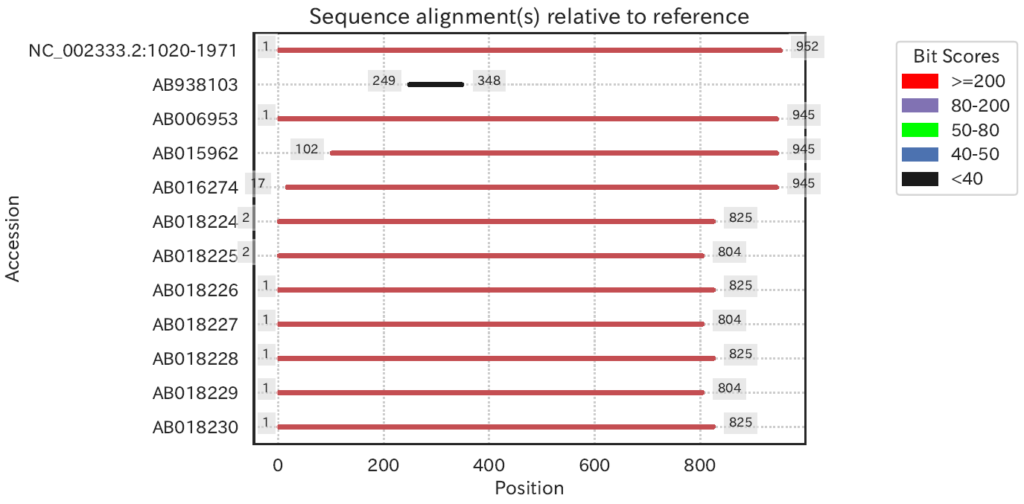
blastnALN.alnpositions.pdf
-rで指定したゼブラフィッシュの12sの配列に対して、-iで指定した配列をblast検索し、得られた出力値がblastnALN.alnpositions.tsvにまとめられます。blastnALN.alnposotions.pdfはそのデータを使ってpythonモジュールのseabornで視覚化してくれます。
簡単にできることをまとめ
Mitohelperのgetrecord関数とgetalignment関数で出来ることをまとめてみます。
- getrecord
- 欲しい魚種名をテキストファイルに記載するとMitofishのデータベースにある配列が取得できる ※ここで出力されたfastaファイルは少し加工が必要
- テキストファイルに指定した種が属する様々な分類階層レベル(Superkingdom ~ Species)の配列を取得できる
- QIIME形式のデータも得られる
- getaglinment
- getrecordで取得した配列に対して、自分が使用したい領域の配列を参照配列とすることで、どのような配列が存在するのかが確認できる
配列を取得したい種を自分で用意してMitoheplerを動かしてみる
ここまででなんとなく使い方が分かってきたので、自分の状況に置き換えて使ってみます。アユを対象としてMiFish領域の配列がMitofish内にあるのか確認してみます。
アユ用のフォルダ(ayuとした)をmitohelperフォルダ上に作成します。
その中にspecies.query.txtとを作成して、その中にPlecoglossus altivelisと書き込みます。
出来たらgetrecord関数を使ってmitohelperを実行します。
|
1 |
$ python3 mitohelper.py getrecord -i ayu/species.query.txt -o ayu/getrecordOUT -d mitofish.all.Feb2021.tsv -l 7 --fasta --taxout |
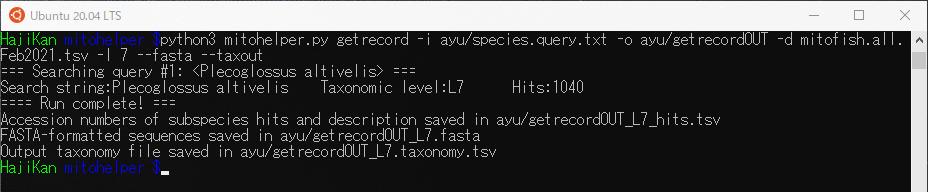
出力されたfastaファイルは”>”毎にうまく改行されていないので、正規表現が使えるテキストエディタなどで”>”毎に改行を加えてfasta形式にします。
これで、Blast検索のクエリ配列が取得できました。
次は参照となる配列を用意します。NCBIに登録されているMiFish領域として切り出された配列を使っていこうと思います。
ayuフォルダの中にAyu.12SMiFish.ref.fastaを作成して、以下の配列情報を記入します。
LC340321.1 Plecoglossus altivelis altivelis mitochondrial gene for 12S rRNA, partial sequence, specimen_voucher: CBM:ZF:16952
Ayu.12SMiFish.ref.fasta
CACCGCGGTTATACGAGTGGCCCAAGTTGAAAGTTACCGGCGTAAAGAGTGGTTAGGGAAACAAAAAACT
AAAGCCGAACACCCTCCAGGCCGTTATACGCTTCTGAGGGCACGAAGCTCCACTACGAAAGTGGCTTTAA
CACACCTGAACCCACGACAACTAAGATA
出来たらgetalignment関数を使ってmitohelperを実行します。
|
1 |
$ python3 mitohelper.py getalignment -i ayu/getrecordOUT_L7.fasta -o ayu/blastnALN -r ayu/Ayu.12SMiFish.ref.fasta --blastn-task blastn |
blastnALN.alnpositions.pdfは以下のように出力されました。
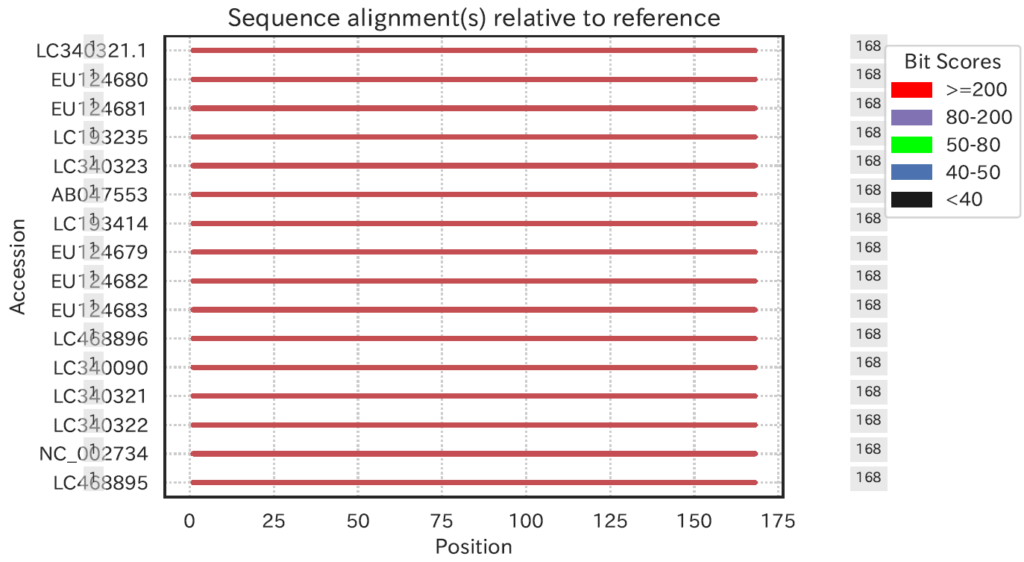
plecoglossus altivelisについてはMiFish領域の配列がちゃんとMitofishに1種類以上は登録されているということはわかります。
欲を言うと塩基のバリエーションも視覚化してくれると嬉しいなとおもいました。
最後に
とりあえず動かしてみましたが、自分の中での使い道があまり見つかりませんでした。パイソンスクリプトの書き方の勉強にはなったので結果オーライです。
最後まで読んで下さりありがとうございます。次は実際に手を動かして得た実験の内容についてお話しできればと考えてます。





