
自分が扱ったことのない分析機器で分析されたデータを解析してみたいなと思うことありませんか?
やっぱり、所属機関にある分析機器にも制限がありますし、ほしいと思ってもなかなか手に入れられないのが普通だと思います。
技術者としてデータ解析は実際に動かしてみたことがあるかどうか、調べてみたことがあるかが一番大切だと思っています。とりあえず環境DNA分析の過程で必要となりそうなものに関して取り組めるようにしておきたいですね。
私が主に扱うのはハイスループットシーケンサー(HTS)から出力されるクオリティスコア付きの塩基配列データです。そして、HTSのデータは基本的に論文掲載時にNCBIやDDBJなどの公共データバンクへ登録する必要があります。
公共データバンクはInternational Nucleotide Sequence Database Collaboration (INSDC) が運用しており、DDBJ (DNA Data Bank of Japan)と欧州のEBI、米国のNCBIによって構成されています。
DDBJ (DNA Data Bank of Japan)は、遺伝研が運営する塩基配列データベース管理拠点で、日本からの登録の99%以上はDDBJを通じて行われています。

上記記事で紹介しているMiFishプライマーの開発論文には、以下のようにDRR030411–030428とあります。
Data accessibility
Custom Ruby scripts used in in silico evaluation of interspecific variation are available from http://dx.doi.org/10.5061/dryad.54v2q. Raw reads from the MiSeq sequencing are available from the DDBJ Sequence Read Archive (DRR030411–030428). The bioinformatic pipeline from data pre-processing through taxonomic assignment (including Perl scripts) is available from http://dx.doi.org/10.5061/dryad.n245j
https://royalsocietypublishing.org/doi/10.1098/rsos.150088
これは、DDBJから発行されるSubmission(DRA), Experiment(DRX), Run(DRR), Analysis(DRZ)のアクセッション番号の一つです。日本語サイトだと以下の記事が分かりやすいです。
DDBJ Sequence Read Archive Handbook
DRAの中にDRXやDRR, DRZの情報が含まれているので、情報を遡っていくとMiFishプライマーの開発論文に関する分析情報はDRA003066というアクセッションナンバーにまとめられているということが分かります。
DRAの番号が分かれば、含まれているFastq.gzファイルや分析の情報などを知ることができます。
この記事の内容
NCBI SRAからFastq.gzファイルをダウンロード
Fastq.gzをDDBJからダウンロードしようとした際、SRAファイルがなかったり、ダウンロードがうまくいかない場合があります。
DDBJのデータはNCBIにもミラーリングされているため、基本的にNCBI SRAからデータを取得するようにしておくと良いと思います。
先ほどの論文のDRA Number(DRA003066)をNCBI SRAで検索して確認してみましょう。
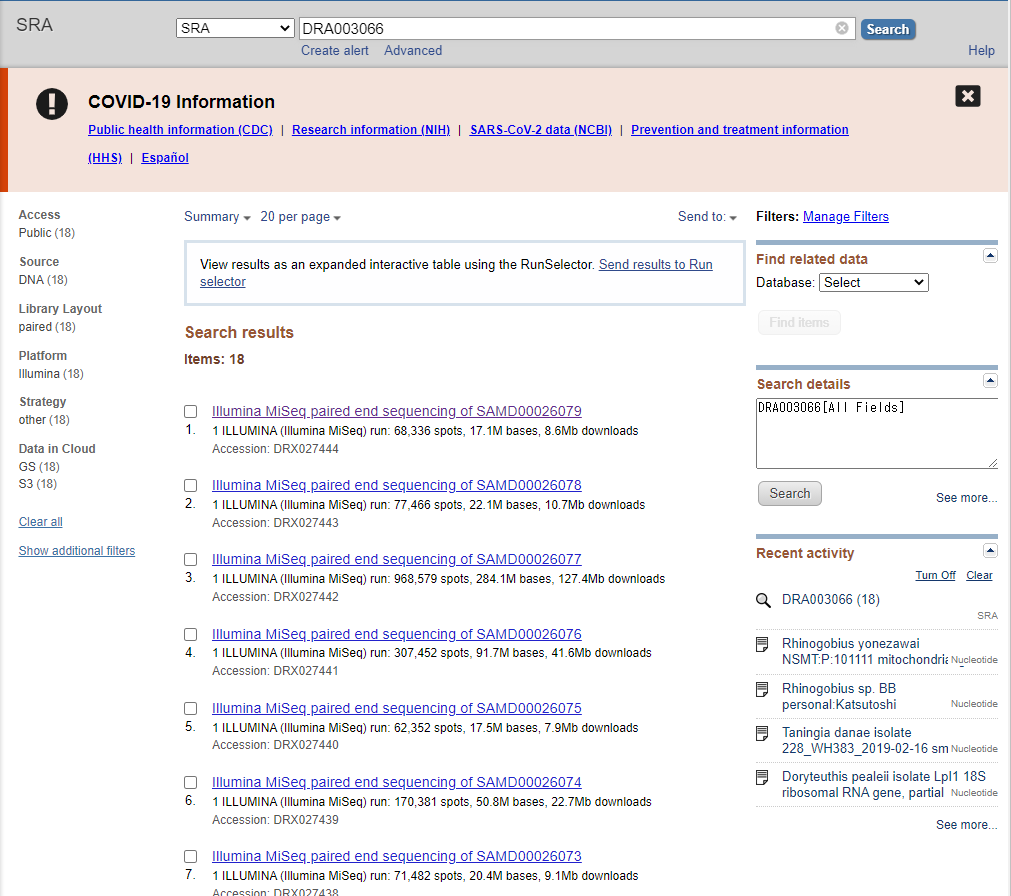
DRR003066の情報は18個に分かれており、1つ目のデータを見てみると以下のような感じになっています。
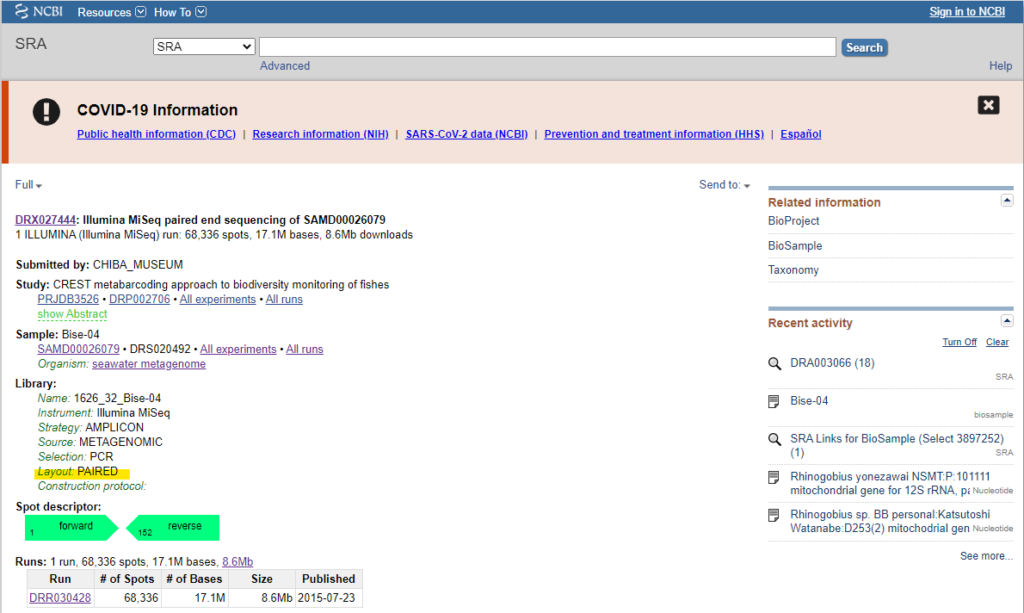
黄色下線部の部分を見るとPAIREDになっているので、このデータはPaired-endシーケンスデータになります。
以降は配列データを扱うprefetchを利用してSRAファイルを取得後、fasterq-dump(fastq-dumpでマルチスレッド処理することで高速化)でFastqファイルへと変換する方法と、直接Fastqファイルをダウンロードする方法を解説していきたいと思います。
動作環境
- windows10
- WSL
- Ubuntu LTS 20.04
- fastq-dump 2.9.3-1
- prefetch 2.9.3-1
SRA Toolkit
SRA Toolkitは公共データバンク(INSDC)のデータを扱うためのツールとライブラリパッケージです。windows, max os, linuxそれぞれに対応しています。
- GitHub repository : https://github.com/ncbi/sra-tools
SRA-Toolkitのインストール
sra-toolkitはaptコマンドで入手することができますが、Ubuntu LTS 20.04ではまだサポートされていないようです。
Ubuntu LTS 18.04以前の方は、
|
1 |
$ sudo apt update -y |
updateコマンドを実行して、パッケージリポジトリを更新し、最新のパッケージ情報を取得してから、
|
1 |
$ sudo apt install sra-toolkit |
でインストールすることができます。
Ubuntu LTS 18.04以降のバージョンの方は、古いファイルをインストールすることで使用できます。
|
1 |
$ sudo apt update -y |
updateコマンドを実行して、パッケージリポジトリを更新し、最新のパッケージ情報を取得しておきます。
もう少し楽な方法が見つかったので更新しました。以下は実行しないでください↓
ダウンロードはLinuxコマンドのwgetを利用します。
|
1 2 3 |
$ wget http://old-releases.ubuntu.com/ubuntu/pool/universe/n/ncbi-vdb/libncbi-vdb2_2.9.3+dfsg-2_amd64.d<kbd>eb</kbd> $ wget http://old-releases.ubuntu.com/ubuntu/pool/universe/n/ncbi-vdb/libncbi-wvdb2_2.9.3+dfsg-2_amd64.deb $ wget http://old-releases.ubuntu.com/ubuntu/pool/universe/s/sra-sdk/sra-toolkit_2.9.3+dfsg-1build2_amd64.deb |
|
1 2 3 |
$ sudo apt install ./libncbi-vdb2_2.9.3+dfsg-2_amd64.deb $ sudo apt install ./libncbi-wvdb2_2.9.3+dfsg-2_amd64.deb $ sudo apt install ./sra-toolkit_2.9.3+dfsg-1build2_amd64.deb |
楽なインストール方法1
SRA-Toolkitをインストールする先のフォルダを作成して、パスを通します。永続的にパスを反映させるのであればホームディレクトリにある.bashrcにパスを記載しておくと楽なのでそこまで解説します。
|
1 2 3 4 |
# sra-toolkitというフォルダをホームディレクトリに作成 $ mkdir -p ~/sra-toolkit # sra-toolkitフォルダに移動 $ cd ~/sra-toolkit |
ここにコンパイル済のパッケージをwgetコマンドでダウンロードします。
|
1 |
$ wget --output-document sratoolkit.tar.gz https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.11.3/sratoolkit.2.11.3-ubuntu64.tar.gz |
※ https://ftp ~ 以降は実行時のsra-toolkitのバージョンに合わせてください。
link : https://github.com/ncbi/sra-tools/wiki/01.-Downloading-SRA-Toolkit
ダウンロードできたら圧縮ファイルをtarコマンドで解凍します。
|
1 |
$ tar -vxzf sratoolkit.tar.gz |
2022年1月現在ではsratoolkit.2.11.3-ubuntu64フォルダが作成されます。この中のbinフォルダに必要な実行コマンドが入っているので、そこにパスを通します。
一時的でいい場合は、
|
1 |
$ export PATH:$PATH:/sra-toolkit/sratoolkit.2.11.3-ubuntu64/bin |
永続的にパスを通しておきたい場合は、nanoコマンドを使用し、.bashrcファイルを編集します。
|
1 |
$ sudo nano ~/.bashrc |
編集モードになったら一番下まで下矢印でいって、下記を記述してください。
|
1 2 |
# Add path export PATH=$PATH:/sra-toolkit/sra-toolkit/sratoolkit.2.11.3-ubuntu64/bin |
編集内容を保存して出る場合は、Ctrl + x → yです。
楽なインストール方法2 mambaを使用したインストール (2023/3/29 更新)
condaに登録されているsra-tookitのバージョンが最新の3.0.2となっていたのでmamba(conda)でインストールします。
|
1 |
mamba install -c bioconda sra-tools -y |
完了したらfasterq-dumpとprefetchのバージョンを確認して、インストールできたか確認してみましょう。
|
1 2 |
$ fasterq-dump -V $ prefetch -V |
2022/1月現在でfasterq-dump: 2.11.3, prefetch: 2.11.2と返ってきました。
(※ 2023/3月現在, どちらも3.0.2でした)
sraファイルをダウンロードするツール : prefetch
開発者githubはこちら。オプションは以下の通り。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 |
Usage: prefetch [options] <SRA accession | kart file> [...] Download SRA or dbGaP files and their dependencies prefetch [options] <SRA file> [...] Check SRA file for missed dependencies and download them prefetch --list <kart file> [...] List the content of a kart file Options: -f|--force <value> force object download one of: no, yes, all. no [default]: skip download if the object if found and complete; yes: download it even if it is found and is complete; all: ignore lock files (stale locks or it is being downloaded by another process: use at your own risk!) -t|--transport <value> transport: one of: fasp; http; both. (fasp only; http only; first try fasp (ascp), use http if cannot download using fasp). Default: both -l|--list list the content of kart file -n|--numbered-list list the content of kart file with kart row numbers -s|--list-sizes list the content of kart file with target file sizes -R|--rows <rows> kart rows to download (default all). row list should be ordered -N|--min-size <size> minimum file size to download in KB (inclusive). -X|--max-size <size> maximum file size to download in KB (exclusive). Default: 20G -o|--order <value> kart prefetch order when downloading kart: one of: kart, size. (in kart order, by file size: smallest first), default: size -a|--ascp-path <ascp-binary|private-key-file> path to ascp program and private key file (asperaweb_id_dsa.putty) --ascp-options <value> arbitrary options to pass to ascp command line -p|--progress <value> time period in minutes to display download progress (0: no progress), default: 1 --eliminate-quals don't download QUALITY column -c|--check-all double-check all refseqs -o|--output-file <FILE> write file to FILE when downloading single file -O|--output-directory <DIRECTORY> save files to DIRECTORY/ -h|--help Output brief explanation for the program. -V|--version Display the version of the program then quit. -L|--log-level <level> Logging level as number or enum string. One of (fatal|sys|int|err|warn|info|debug) or (0-6) Current/default is warn -v|--verbose Increase the verbosity of the program status messages. Use multiple times for more verbosity. Negates quiet. -q|--quiet Turn off all status messages for the program. Negated by verbose. --option-file <file> Read more options and parameters from the file. -+|--debug <Module[-Flag]> Turn on debug output for module. All flags if not specified. prefetch : 2.9.3 ( 2.9.3-1 ) |
prefetchはSRAデータをダウンロードしてくるコマンドです。一度.sraファイルをダウンロードしておくとfastqへの変換に失敗してしまったりした際にやり直しが楽です。
DRR030411–030428 のRunデータがまとめられているので、それらを一括でダウンロードします。
まず、DRR030411- 030428までをリストにしたテキストファイルを用意します(以下のファイルを使っていただいても結構です)。
ダウンロードしたいRunデータのリスト(SRR_Acc_list.txt)が用意できたら、下記コマンドを実行して、.sraファイルをダウンロードします (10分くらい 計約4GB) 。
|
1 |
$ prefetch --output-directory ./ --option-file SRR_Acc_list.txt |
./の部分は例えばResultsフォルダなどを作ってもらって./Resultsにするとそのフォルダにファイルが保存されます。
また、listファイルを作成したりするのが面倒で、DRRの番号が連番なら以下のようなコマンドで同様の結果を得ることもできます。
|
1 |
$ for i in `seq 11 28` ; do prefetch DRR0304$i --output-directory ./ ; done |
次はfastqerq-dumpを使ってダウンロードした.sraファイルからFastqファイルを生成してみましょう。
sraファイルからfastqファイルを生成するツール : fasterq-dump
開発者githubはこちら。オプションは以下の通り。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 |
Usage: fasterq-dump <path> [options] Options: -o|--outfile output-file -O|--outdir output-dir -b|--bufsize size of file-buffer dflt=1MB -c|--curcache size of cursor-cache dflt=10MB -m|--mem memory limit for sorting dflt=100MB -t|--temp where to put temp. files dflt=curr dir -e|--threads how many thread dflt=6 -p|--progress show progress -x|--details print details -s|--split-spot split spots into reads -S|--split-files write reads into different files -3|--split-3 writes single reads in special file --concatenate-reads writes whole spots into one file -Z|--stdout print output to stdout -f|--force force to overwrite existing file(s) -N|--rowid-as-name use row-id as name --skip-technical skip technical reads --include-technical include technical reads -P|--print-read-nr print read-numbers -M|--min-read-len filter by sequence-len --table which seq-table to use in case of pacbio --strict terminate on invalid read -B|--bases filter by bases -h|--help Output brief explanation for the program. -V|--version Display the version of the program then quit. -L|--log-level <level> Logging level as number or enum string. One of (fatal|sys|int|err|warn|info|debug) or (0-6) Current/default is warn -v|--verbose Increase the verbosity of the program status messages. Use multiple times for more verbosity. Negates quiet. -q|--quiet Turn off all status messages for the program. Negated by verbose. --option-file <file> Read more options and parameters from the file. -+|--debug <Module[-Flag]> Turn on debug output for module. All flags if not specified. fasterq-dump : 2.9.3 ( 2.9.3-1 ) |
基本的に環境DNAメタバーコーデイングのシーケンスデータはpaired-endで読まれていることが多いです。Illuminaシーケンサーでのpaired-end ampliconシーケンスは解読したい150bp ~ 500bp程度の領域の配列を150bp ~ 300bpずつ解読できるようなキットで両側から配列を読んで、オーバーラップしている部分を繋ぎ合わせて配列決定するような方法です。
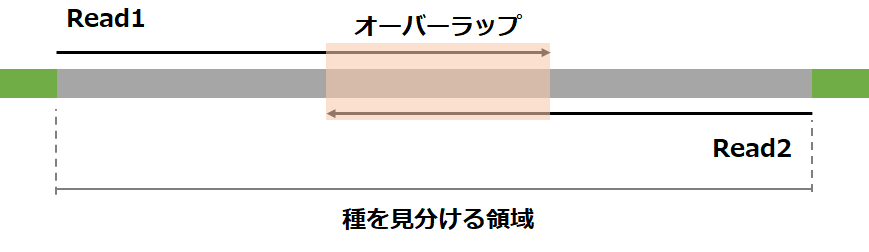
single-endシーケンスと比べて以下のように長い配列長で読むことができるのが特徴です。
.sraファイルからFastqファイルを取得
ダウンロードした.sraファイルをfastqファイルに変換します。
|
1 |
$ for i in `seq 11 28` ; do fasterq-dump -p -e 24 DRR0304$i.sra ; done |
1行のコマンドで変換は終了です。
オプションを2つ使用しています。-pは進捗状況を表示、-eは複数のコアを使用しての並列処理オプションです。*2022/2/23 追加
_1.fastqと_2.fastq はイルミナのHTSから出力された状態だとR1, R2にあたるファイルで、fowardプライマーとReverseプライマーで読まれた配列になります。
- DRR0304XX.sra_1.fastq
- DRR0304XX.sra_2.fastq
.fastqファイルはgzipで圧縮して名前を_1 →_R1、_2 → _R2に変換しておきます。
|
1 2 |
$ for i in `seq 11 28 ` ; do gzip -k -v -c DRR0304$i.sra_1.fastq > DRR0304$i.sra_R1.fastq.gz; done $ for i in `seq 11 28 ` ; do gzip -k -v -c DRR0304$i.sra_2.fastq > DRR0304$i.sra_R2.fastq.gz; done |
これで、それぞれの.sraファイルのfastqファイルを取得することができました。
paired-end sequenceをfasterq-dumpでダウンロード
.sraファイルからfastqファイルを作成するのに使用したfasterq-dumpは直接SRAから.fastqファイルをダウンロードすることもできます。その際、注意点としてpaired-endでシーケンスされた.sraデータは--split-filesオプションを使用しないと、つながった状態のものがダウンロードされるようです。
そのため、分割してR1とR2ファイルをダウンロードするには--split-filesオプションを使用します。DRA003066の中にあるFastqファイルはDRR030411–030428と複数ファイルに分かれて登録されているので、forを使ってそれぞれのファイルをダウンロードします。
|
1 |
$ for i in `seq 11 28` ; do fasterq-dump -p -e 24 DRR0304$i --split-files ; done |
これでDRR0030XXのXXの部分を11から28まで変化させて自動的にファイルをダウンロードすると以下のようなファイル名のFastqファイルが出力されます。
- DRR0304XX_1.fastq
- DRR0304XX_2.fastq
fastqファイルがダウンロードできればprefetchの章でも解説したように、圧縮しつつファイル名を変更しておきます。
|
1 2 |
$ for i in `seq 11 28 ` ; do gzip -k -v -c DRR0304$i_1.fastq > DRR0304$i_R1.fastq.gz ; done $ for i in `seq 11 28 ` ; do gzip -k -v -c DRR0304$i_2.fastq > DRR0304$i_R2.fastq.gz ; done |
まとめスクリプト
prefetchを使用して、.sraファイルをダウンロードしたのちに_R1.fastq.gz, _R2.fastq.gzファイルを取得するパターン。
.sraファイルはsraフォルダを、fastqファイルはfastqフォルダを、.fastq.gzはfastqgzフォルダを作成してそこに保存するようにしました。
また、以下はアクセッションナンバーのリストが記載されたtxtファイルを用意していない前提のスクリプトです。
|
1 2 3 4 5 6 7 8 9 10 11 12 |
#フォルダの作成 $ mkdir sra fastq #.sraファイルのダウンロード $ for i in `seq 11 28` ; do prefetch DRR0304$i --output-directory ./sra ; done #fastqファイルを作成 $ for i in `seq 11 28` ; do fasterq-dump -p -e 24 ./sra/DRR0304$i.sra --outdir ./fastq ; done #fastqファイルを圧縮して名前を変更 $ for i in `seq 11 28 ` ; do gzip -k -v -c ./fastq/DRR0304$i.sra_1.fastq > ./fastqgz/DRR0304$i.sra_R1.fastq.gz ; done $ for i in `seq 11 28 ` ; do gzip -k -v -c ./fastq/DRR0304$i.sra_2.fastq > ./fastqgz/DRR0304$i.sra_R2.fastq.gz ; done |
以下は、fastqerq-dumpを使ってSRAから直接fastqファイルをダウンロードするまとめスクリプトです。
|
1 2 3 4 5 6 7 8 9 |
#フォルダの作成 $ mkdir fastq #fastqファイルのダウンロード $ for i in `seq 11 28` ; do fasterq-dump -p -e 24 DRR0304$i --split-files ; done #fastqファイルを圧縮して名前を変更 $ for i in `seq 11 28 ` ; do gzip -k -v -c DRR0304$i_1.fastq ; DRR0304$i_R1.fastq.gz ; done $ for i in `seq 11 28 ` ; do gzip -k -v -c DRR0304$i_2.fastq ; DRR0304$i_R2.fastq.gz ; done |
オプション : PMiFish piplineで解析してみる
PMiFish piplineについては以下の記事を参考に進めてもらえればと思います。

MiFishプライマーの開発論文では、MiFish-UとMiFish-Eの2種類のプライマーセットを使用しているので、PMiFish piplineのDatabaseフォルダにあるPrimer.txtは以下のように記載します。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
#Primer pairs (Forward and Reverse) need to be the same name #Primer name can not use space. Use "_" or "-" instead of space #If a user put "#" at beginning of line, the primer would be used in the analysis Forward primers #MiFish-U NNNNNNGTCGGTAAAACTCGTGCCAGC #MiFish-E NNNNNNGTTGGTAAATCTCGTGCCAGC #MiFish-tuna NNNNNNATGTCCTTCCTCCTTATCGGCTG Reverse primers #MiFish-U NNNNNNGTTTGACCCTAATCTATGGGGTGATAC #MiFish-E NNNNNNGTTTGATCCTAATCTATGGGGTGATAC #MiFish-tuna NNNNNNTTGCCAGTGGCAGCTACGATC |
複数のプライマーを使用しているので、setting.txtのDivideはNo→Yesに変更しておきます。
あとはそのままの設定でもOKです。パイプラインを動かして結果を出力してみましょう。
これ以降の解析についてはまた別記事で解説したいと思います。最後まで読んでくださりありがとうございます。
番外編 : pysradbを使ってSRP(研究ごとに割り振られる番号)の情報をもとに配列データを取得する

pysradbはSRA/ENA/GEOからメタデータを取得するためのPythonパッケージです。
mamba使ったインストールとは以下の通り。
|
1 |
mamba install -c bioconda pysradb -y |
ハイスループットシーケンサーの配列データに紐づくメタデータは、サマリー情報を持つSRAも含め、
- 研究やプロジェクトに関する情報を持つ : “SRP”
- 機器やライブラリといった実験に関する情報を持つ : “SRX”
- 塩基配列と対応するクオリティスコアの情報を持つ : “SRR”
が入れ子構造でまとまっています。
つまり研究やプロジェクトごとに付与されるSRPの番号を使うことで、その研究で解読された配列データや関連する情報を一気に取得することができるようになります。
pysradbの便利なのはgrepと組み合わせることで、取得したメタデータの情報を使ってフィルタリング後、欲しい配列データをMulti threadでダウンロードできる点です。
例えば、SRP420753は2023年にOceanOmicsがRowley Shoals周辺の環境水から12S, 16S, COIのアンプリコンとWGSの塩基配列を決定した配列データがまとめられています。
以下のコマンドでSRP420753に属する266サンプル分のメタデータを取得できます(1分ほど)。
|
1 |
pysradb metadata SRP420753 --detailed > SRP420753.tsv |
この中で12S(=MiFish)の配列だけほしいな。と思った時には、以下のようにfasterq-dumpと組み合わせることでさくっと配列データ(fastq.gz)を得ることが出来ます。
|
1 2 3 4 |
mkdir 00sra pysradb metadata SRP420753 --detailed | grep 'MiFish' | pysradb download -y -t 32 --out-dir ./00sra find . -name '*.sra' -execdir fasterq-dump -p -e 32 {} \; find . -name '*.fastq' -execdir pigz -p 32 {} \; |
簡単に説明すると、MiFishというワードでフィルタリングをかけて、合致したSRRのdownloadオプションで配列取得し、fastereq-dumpでsra→fastqに変換後、pigzで並列圧縮しています。
別のSRP番号で実施するときも、一度SRPに対応するデータ一覧を見て、grepの部分に記載するフィルタリングワードを決めてから実行するのが手間を少なく欲しい配列データを取得することができる一番の近道だと思います。
最後に、pysradbのオプションは以下の通りです。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 |
usage: pysradb [-h] [--version] [--citation] {metadata,download,search,gse-to-gsm,gse-to-srp,gsm-to-gse,gsm-to-srp,gsm-to-srr,gsm-to-srs,gsm-to-srx,srp-to-gse,srp-to-srr,srp-to-srs,srp-to-srx,srr-to-gsm,srr-to-srp,srr-to-srs,srr-to-srx,srs-to-gsm,srs-to-srx,srx-to-srp,srx-to-srr,srx-to-srs} ... pysradb: Query NGS metadata and data from NCBI Sequence Read Archive. version: 2.1.0. Citation: 10.12688/f1000research.18676.1 optional arguments: -h, --help show this help message and exit --version show program's version number and exit --citation how to cite subcommands: {metadata,download,search,gse-to-gsm,gse-to-srp,gsm-to-gse,gsm-to-srp,gsm-to-srr,gsm-to-srs,gsm-to-srx,srp-to-gse,srp-to-srr,srp-to-srs,srp-to-srx,srr-to-gsm,srr-to-srp,srr-to-srs,srr-to-srx,srs-to-gsm,srs-to-srx,srx-to-srp,srx-to-srr,srx-to-srs} metadata Fetch metadata for SRA project (SRPnnnn) download Download SRA project (SRPnnnn) search Search SRA/ENA for matching text gse-to-gsm Get GSM for a GSE gse-to-srp Get SRP for a GSE gsm-to-gse Get GSE for a GSM gsm-to-srp Get SRP for a GSM gsm-to-srr Get SRR for a GSM gsm-to-srs Get SRS for a GSM gsm-to-srx Get SRX for a GSM srp-to-gse Get GSE for a SRP srp-to-srr Get SRR for a SRP srp-to-srs Get SRS for a SRP srp-to-srx Get SRX for a SRP srr-to-gsm Get GSM for a SRR srr-to-srp Get SRP for a SRR srr-to-srs Get SRS for a SRR srr-to-srx Get SRX for a SRR srs-to-gsm Get GSM for a SRS srs-to-srx Get SRX for a SRS srx-to-srp Get SRP for a SRX srx-to-srr Get SRR for a SRX srx-to-srs Get SRS for a SRX |
参考
- NCBI SRA から FASTQ をダウンロードする方法
- DDBJ Sequence Read Archive
- SRA-toolkitを使ってサンプルデータをダウンロード
- SRA/DRA
- SRA toolkit for Ubuntu 20.04 LTS
- SRA Toolkitの使い方 ~ インストールと設定 ~
- Install sra toolkit on ubuntu20.04





