
この記事の内容
やりたいこと
Entrezによるデータベース検索システムを使って配列情報を取得します。
Entrezの検索システムは皆さんがNCBIで配列を検索したりするときに使うこのページそのものだと思っていただいたらいいと思います。
注意点
情報を取得する際、サーバーに大きな負荷がかからないようにガイドラインに記載されているルールは守る。
簡単にまとめると以下の通りです。2と3についてはBiopythonが勝手にやってくれるそうです。
- 100以上のリクエストを送る場合は、週末かピークタイム以外の時間に行う
- 通常のアドレスではなく、http://eutils.ncbi.nlm.nih.gov/ を使用する
- リクエストの頻度を3リクエスト/1s 以下になるようにする
- e-mailとE-utilities APIの設定はしておく方がいい
(APIを登録しておくとリクエスト制限が緩和される)
また、本記事はJupyter lab上でスクリプトを動かして、markdwon記法で記事を書いています。
Rstudioのような開発環境で、コードを実行すると結果がすぐわかるのはすごく使い勝手がよかったです。
参考
- Biopython を利用したNCBIのEntrez データベースへのアクセス
- biopythonでNCBIのnuccoreデータベースからDNAデータをとってくる
- The E-utilities In-Depth: Parameters, Syntax and More
- Entrez efetchで配列レコードを大量取得する
- Biopythonを使ってPMCから論文取得
大変勉強になる記事ばかりでした。ここで御礼申し上げます。
動作環境
- Ubuntu 20.04 LTS
- python 3.8.5
- biopython 1.78
事前準備
Setting textファイルを作成
ディレクトリにSetting.txtを作成します。毎回パラメーターを記入するのは面倒なので、setting.txtに以下の情報をまとめて記入して、都度必要になったら呼び出すという形にします。
setting.txtの記載内容
emailアドレスとAPI keyは自身のものを記載してください。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 |
####Caution#### All file names must not include space. Use "_" or "-" instead of space. ############### # Setting values about all scripts. Email = Your e-mail adress Api_key = Your API key # ==== Setting values about Download ID === ## Variables for efetch Database1 = nucleotide # Select the database to use. Rettype1 = fasta # Select the format to download files. Retmode1 = text # Select the file extension to download data. Count_in_query1 = 20 # How many queries to request at a time. Setting range is 1 to 100000 at a time. minday = 2000/01/01 Species = Plecoglossus altivelis Genetic = mitochondrion |
NCBIへの自己紹介
|
1 2 3 4 |
#emailアドレス Email = "自分のメールアドレス" #Apiキー Api_key = "NCBIでアカウント登録した時に発行されたAPI番号" |
emailやAPI keyは人によって異なる部分です。
パラメーター
添付画像の情報を指定していると思っていただけたらいいと思います。
|
1 2 |
#検索するデータベース Database = "nucleotide" # ヌクレオチドのデータベースを使用 |
|
1 2 |
# データをダウンロードするときのフォーマット Retmode = text # テキストで出力 |
RetmodeとRettypeの適切な組み合わせについてはこのページを参照ください。
|
1 2 |
# 1回の要求でダウンロードするデータ数 Count_in_query = 20 |
|
1 2 3 4 5 6 |
#いつ以降の配列を取得したいか minday = 2000/01/01 # いつまでの配列を取得したいか # 実行日を指定したい場合はスクリプト内に記載する為記載不要 maxday = 2021/01/27 |
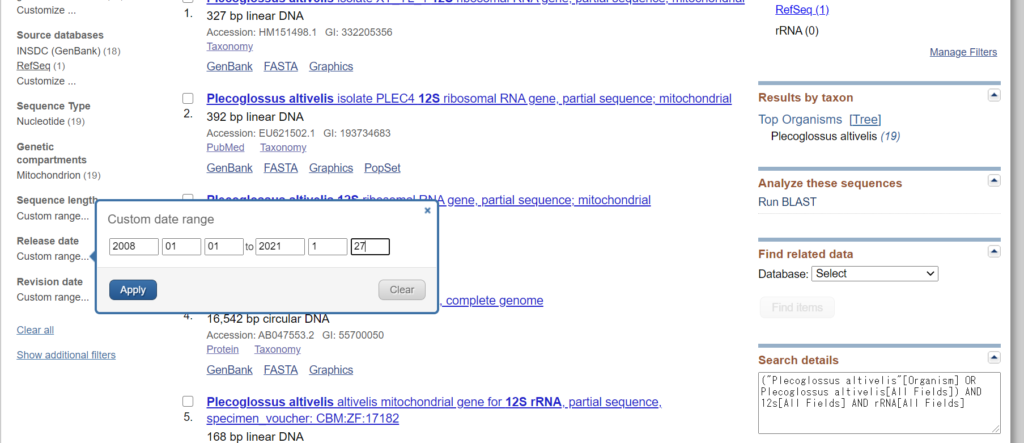
|
1 2 3 4 |
# 対象種を指定 species = Plecoglossus altivelis # 対象遺伝子を設定 genetic = mitochondrion |
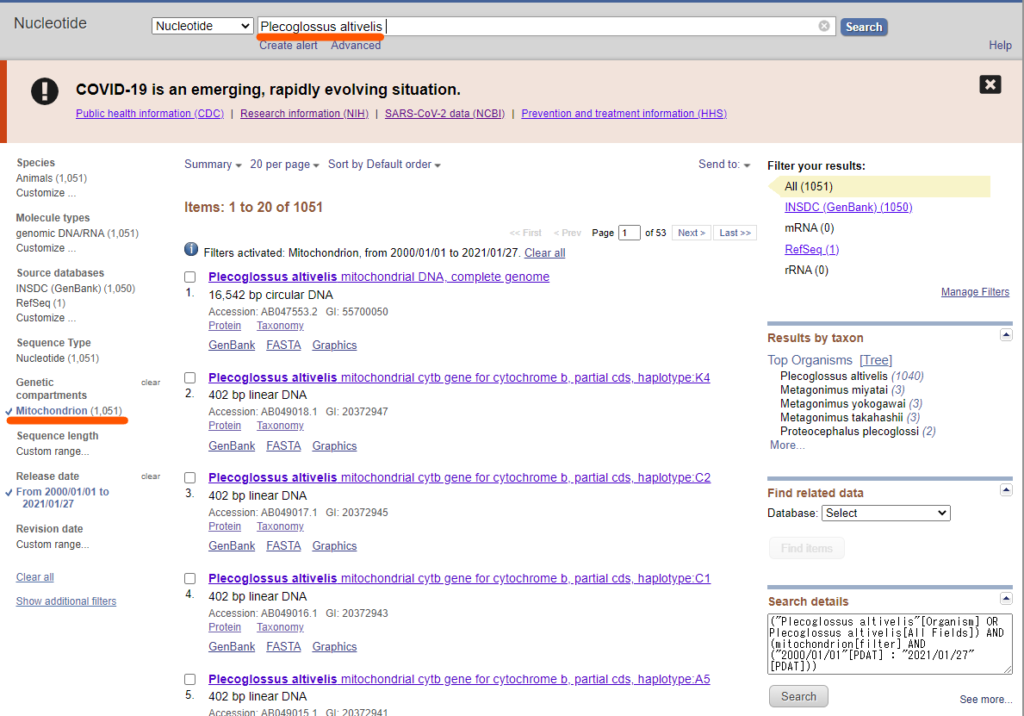
今回はアユ(Plecoglossus altivelis)のミトコンドリア遺伝子を指定します。
0. 全体の設定
0-1. import module
|
1 2 3 4 5 6 7 8 9 10 11 12 13 |
import subprocess as sp import pandas as pd import pathlib, glob, re, pprint, os, datetime, shutil, time, sys from Bio import Entrez, SeqIO # from Bio.GenBank.Record import Record from Bio.SeqFeature import SeqFeature, FeatureLocation from Bio.SeqRecord import SeqRecord from datetime import timedelta from time import sleep from dateutil.relativedelta import relativedelta from subprocess import Popen, PIPE from tqdm import tqdm from termcolor import colored, cprint |
0-2. 結果をまとめるフォルダーのチェック
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 |
def check_results_folder(): # Reusltsフォルダの有無のチェック if pathlib.Path("./Results").is_dir() == True: shutil.rmtree("./Results") # フォルダを消して pathlib.Path("./Results").mkdir(exist_ok=True) # 作成 else: pathlib.Path("./Results").mkdir(exist_ok=True) # IDをまとめるファイルのチェック if pathlib.Path('./Results/ID.txt').is_file == True: pathlib.Path("./Results/ID.txt").unlink() else: with open('./Results/ID.txt', mode='w') as i: i.write("") # Fastaファイルをまとめるファイルのチェック if pathlib.Path("./Results/Fasta").is_dir() == False: pathlib.Path("./Results/Fasta").mkdir(exist_ok=True) else: pass # チェック check_results_folder() |
0-3. プログレスバーの色の設定
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 |
# Color setting class Color: BLACK = '\033[30m' RED = '\033[31m' GREEN = '\033[32m' YELLOW = '\033[33m' BLUE = '\033[34m' PURPLE = '\033[35m' CYAN = '\033[36m' WHITE = '\033[37m' END = '\033[0m' BOLD = '\038[1m' UNDERLINE = '\033[4m' INVISIBLE = '\033[08m' REVERCE = '\033[07m' |
0-4. setting.txtの読み込み
|
1 2 |
with open('Setting.txt', encoding = "utf-8") as st: txt = st.read() |
0-5. NCBIへ自己紹介
reモジュールを使ってアドレスとAPI keyを取得します。
|
1 2 |
Entrez.email = re.search(r'Email\s*=\s*(\S+)', txt).group(1) Entrez.api_key = re.search(r'Api_key\s*=\s*(\S+)', txt).group(1) |
1. Genbank IDの取得
1-1. 使用する変数の設定
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
# データベースを取得 db = re.search(r'Database1\s*=\s*(\S+)', txt).group(1) # ファイルをダウンロードするときの形式 rettype = re.search(r'Rettype1\s*=\s*(\S+)', txt).group(1) # データをダウンロードするときのフォーマット retmode = re.search(r'Retmode1\s*=\s*(\S+)', txt).group(1) # 1回の要求でダウンロードするデータ数 count_in_query = re.search(r'Count_in_query1\s*=\s*(\S+)', txt).group(1) # いつからのデータを取得するか minday = re.search(r'minday\s*=\s*(\S+)', txt).group(1) # いつまでのデータを取得するか (常に今日になるようになっています) maxday = datetime.date.today().strftime("%Y/%m/%d") # 対象種を取得 species = re.search(r'Species\s*=\s*(.*)', txt).group(1) # 対象遺伝子を設定 genetic= re.search(r'Genetic\s*=\s*(\S+)', txt).group(1) |
1-2. esearchを動かす
esearchはEntrezの検索機能の部分で、efetchはダウンロード機能の部分です。IDを検索して取得したいのでesearchを使います。
|
1 2 3 4 5 6 |
# 配列情報の検索 handle_search = Entrez.esearch(db = db, retmax = count_in_query, term = f'{species}[Organism] AND {genetic}[filter]') record_search = Entrez.read(handle_search) # 取得したIDのList SeqID = record_search["IdList"] |
retmaxを20と指定したので、検索上位20本分の配列情報に付与されたGenbank IDを取得することが出来ました。count_in_queryの数値は1~100000まで指定できるので、もっと大量の情報が欲しい時はこの数値を調整してみてください。
1-3. IDをテキストファイルに書き出す
|
1 2 3 4 5 6 |
# 操作しやすいようにID毎に改行する str_ID = '\n'.join(SeqID) # ResultsフォルダのID.txtに取得したIDを書き出す with open('./Results/ID.txt', mode = 'a') as f: f.write(str_ID + "\n") |
これで、2000年1月1日以降に登録されたアユのミトコンドリアDNA配列に付与されているGenbankIDが取得できました。
次は取得したGenbankIDをもとにそれぞれの領域情報と、配列を取得していきます。
ResultsフォルダのID.txtには先ほど取得した20本分のGenbank IDが記載されています。それらを使ってそれぞれのIDの登録配列のDNA領域の情報を取得してみましょう。
|
1 2 |
# IDが記載されているID.txtの読み込み ID_dataframe = pd.read_csv("./Results/ID.txt", encoding="UTF-8", header=None) |
2-1. 対象IDの配列を遺伝子領域ごとにFastaファイルに出力する
対象種はアユ、対象ゲノムはミトコンドリアDNAで検索した時にヒットした20個分のデータのGenbank IDがID.txtに格納されています。
ミトコンドリアDNAはリボソームRNAやtransfer RNAなど数十種類の遺伝子を有しています。MiFishプライマーなどは12s rRNA遺伝子に設計されていたり、系統解析に使われるようなシトクロムb遺伝子はミトコンドリアDNAにコードされています。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 |
# New function define def makeSeqRecord(seq_obj, id_for_fasta): record = SeqRecord(seq_obj, id=id_for_fasta, description="") return record # rRNA or CDS or tRNA misc_feature feature_str = 'rRNA' # Genbank ID数を取得 id_num = len(ID_dataframe) # Genbank ID の数だけForで情報を取得する for i in tqdm(range(0, id_num), desc = Color.CYAN + 'Progress rate' + Color.END): # IDの配列情報をGenbank形式で取得 handle_info = Entrez.efetch(db = db, id = ID_dataframe.iloc[i,0], rettype = 'gb', retmode = 'text') for record_info in SeqIO.parse(handle_info, "genbank"): # gbを指定するとGenbank形式の情報を取得する # リスト格納 record_list = [] # 配列全体を格納 parent_sequence = record_info.seq # 学名を格納 organism = record_info.annotations['organism'] # Accession numberを格納 acc = record_info.id # 各領域ごとに配列を切り分けて配分 for feature in record_info.features: # 不明な遺伝子領域 misc_feature 以外の場合 if feature.type != 'misc_feature' and feature_str == feature.type: sequence_slice = feature.location.extract(parent_sequence) # 配列の末端取得 strand_int = feature.location.strand if strand_int == 1: strand = 'top' # 5'末端 elif strand_int == -1: strand = 'btm' # 3'末端 else: strand = 'cannotbe' # 配列の開始位置指定 start = feature.location.parts[0].start.position + 1 # 配列の終了位置指定 end = feature.location.parts[-1].end.position # 遺伝子領域の情報取得 if 'gene' in feature.qualifiers: gene = feature.qualifiers['gene'][0] else: gene = 'NA' # 転写産物?の情報取得 if 'product' in feature.qualifiers: product = feature.qualifiers['product'][0] else: product = 'NA' # Fasta形式にした時の情報部分(> ~~~ の部分)の作成 id_line = acc + '_' + organism + '_' + gene + '_' + product + '_' id_line += ' (' + str(start) + '..' + str(end) + ', strand=' + strand + ')' # レコードから取得した情報をFasta形式にする record_list = makeSeqRecord(sequence_slice, id_line) # Fastaファイルの出力 if feature_str != 'D-loop': fasta_file_name = './Results/Fasta/' + acc + f'_{product}_' + feature.type + '.fasta' else: fasta_file_name = './Results/Fasta/' + acc + f'_{feature_str}_test.fasta' # 一応0以上かチェック type_count = len(record_list) with open(fasta_file_name, mode = 'w') as ffn: if type_count > 0: SeqIO.write(record_list, ffn, 'fasta') else : ffn.write('No Annotation Found') # merge fasta file name mfa = f'./Results/Fasta/mergefa.fas' with open(mfa, mode = 'a') as mfa: if type_count > 0: SeqIO.write(record_list, mfa, 'fasta') else: pass with open("./Results/list.txt", mode = 'w') as spl: spl.write(organism + '\n') # 不明な遺伝子領域 misc_feature の場合(D-loopがここに含まれる場合があるため作成) elif feature_str == 'misc_feature' and feature_str == feature.type or feature_str == 'misic_feature' and re.search('(d|D)-(l|L)oop', feature.type) == True: # feature.qualifiers フィールドにD-loopの記載がある場合があるため if 'note' in feature.qualifiers: note = feature.qualifiers['note'][0] else: note = 'No Annotation' sequence_slice = feature.location.extract(parent_sequence) # 配列の末端 strand_int = feature.location.strand if strand_int == 1: strand = 'top' # 5'末端 elif strand_int == -1: strand = 'btm' # 3'末端 else: strand = 'cannotbe' # 配列の開始位置 start = feature.location.parts[0].start.position + 1 # 配列の終了位置 end = feature.location.parts[-1].end.position # 転写産物?の情報 product = 'NA' # Fasta形式にした時の情報部分の作成 id_line = acc + '_' + organism + '_' + note + '_' id_line += ' (' + str(start) + '..' + str(end) + ', strand=' + strand + ')' # レコードから取得した情報をFasta形式にする record_list = makeSeqRecord(sequence_slice, id_line) # Fastaファイルの出力 fasta_file_name = './Results/Fasta/' + acc +'_' + feature.type + '.fasta' type_count = len(record_list) with open(fasta_file_name, mode = 'w') as ffn: if type_count > 0: SeqIO.write(record_list, ffn, 'fasta') else : ffn.write('No Annotation Found') # merge fasta file name mfa = './Results/Fasta/mergefa.fas' with open(mfa, mode = 'a') as mfa: if type_count > 0: SeqIO.write(record_list, mfa, 'fasta') else: pass # リストに追加された種名を記入 add_organism_list = [] add_organism_list = add_organism_list.append(organism) print(add_organism_list) with open("./Results/list.txt", mode = 'w') as spl: spl.write(organism + '\n') else: pass |
以下のフォルダ構成になります。1本しかhitしてないのでmergefa.fasには1本しかないですが、対象種や対象feature、retmaxを変えたりしたら複数配列が取得できます。
- Fasta
- LC600708.1_12S ribosomal RNA_rRNA.fasta
- mergefa.fas
- ID.txt
- list.txt
2-2.DNA領域に構わず、対象IDの配列を一括取得する
領域ごとに切り出すとかファイルを分けるとかせずに、IDに対応する配列を全部取得するんだ。という方はこちらを使用してみてください。2-1の部分をごっそり以下のコードに変更します。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 |
# New function define def makeSeqRecord(seq_obj, id_for_fasta): record = SeqRecord(seq_obj, id=id_for_fasta, description="") return record # IDが記載されているID.txtの読み込み ID_dataframe = pd.read_csv("./Results/ID.txt", encoding="UTF-8", header=None) # Genbank ID数を取得 id_num = len(ID_dataframe) # GWnbank ID の数だけForで情報を取得する for i in tqdm(range(0, id_num), desc = Color.CYAN + 'Progress rate' + Color.END): handle = Entrez.efetch(db = db, id = ID_dataframe.iloc[i,0], rettype = 'gb', retmode = 'text') for record_info in SeqIO.parse(handle, "genbank"): # gbを指定するとGenbank形式の情報を取得する # リスト格納 record_list = [] # 配列全体を格納 parent_sequence = record_info.seq # 学名を格納 organism = record_info.annotations['organism'] # Accession numberを格納 acc = record_info.id # 各領域ごとに配列を切り分けて配分 for feature in record_info.features: # 不明な遺伝子領域 misc_feature 以外の場合 if feature.type == 'source': sequence_slice = feature.location.extract(parent_sequence) # 配列の末端取得 strand_int = feature.location.strand if strand_int == 1: strand = 'top' # 5'末端 elif strand_int == -1: strand = 'btm' # 3'末端 else: strand = 'cannotbe' # 配列の開始位置指定 start = feature.location.parts[0].start.position + 1 # 配列の終了位置指定 end = feature.location.parts[-1].end.position # Fasta形式にした時の情報部分(> ~~~ の部分)の作成 id_line = acc + '_' + organism + '(' + str(start) + '..' + str(end) + ', strand=' + strand + ')' # レコードから取得した情報をFasta形式にする record_list = makeSeqRecord(parent_sequence, id_line) # アクセッションナンバー.fa のファスタファイルでファイル名を指定 fasta_file_name = './Results/Fasta/' + record_info.id + '.fasta' # 一応0以上かチェック type_count = len(record_list) # 書き出し with open(fasta_file_name, mode = 'w') as ffn: if type_count > 0: SeqIO.write(record_list, ffn, 'fasta') else : ffn.write('No Annotation Found') # merge fasta file name mfa = './Results/Fasta/mergefa.fas' with open(mfa, mode = 'a') as mfa: if type_count > 0: SeqIO.write(record_list, mfa, 'fasta') else: pass |
以下がResultsフォルダ内の構成になります。mergefa.fasには20本分のfastaファイルが一つにまとめられています。
- Fasta
- Fastaファイル20本
- mergefa.fas
- ID.txt
場合に応じて使い分けてみてください。
番外編
2-2で取得した配列の特徴をmatplotlibで視覚化してみる。
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 |
# 成型 with open('./Results/Fasta/mergefa.fas', mode = 'r') as fa: if i == 0: fasta = fa.read() fasta_rp = re.sub(r'>', r'\n>', fasta) fasta_rp = re.sub(r"top\)\n", r"top)\t", fasta_rp) fasta_rp = re.sub(r"btm\)\n", r"btm)\t", fasta_rp) fasta_rp = re.sub(r'([A-Z]|-$)\n', r'\1', fasta_rp) with open('./Results/Fasta/mergefa_edit.fas', mode = "w") as wr: wr.write(fasta_rp) else: fasta = fa.read() fasta_rp = re.sub(r'>', r'\n>', fasta) fasta_rp = re.sub(r'^\n', '', fasta_rp) fasta_rp = re.sub(r"top\)\n", r"top)\t", fasta_rp) fasta_rp = re.sub(r"btm\)\n", r"btm)\t", fasta_rp) fasta_rp = re.sub(r'([A-Z]|-$)\n', r'\1', fasta_rp) # 書き出し with open('./Results/Fasta/merge_edit.fas', mode='w') as wr: wr.write(fasta_rp) # テーブルデータとして読み込み mfa = pd.read_csv('./Results/Fasta/merge_edit.fas', encoding="UTF-8", header = None, index_col = False, sep = "\t") # 列名を変更 mfa = mfa.rename(columns={0:"ID",1:"seq"}) # 配列の長さを算出 mfa['seq_len'] = mfa['seq'].str.len() |
|
1 2 3 4 5 6 7 8 9 10 |
# 配列長でhistグラムを描画してみる import matplotlib.pyplot as plt from pylab import rcParams rcParams['figure.figsize'] = 20, 5 rcParams['font.size'] = 25 mfa.hist(bins=50) plt.tight_layout() plt.show() |

まとめのコードなど
いくつかのパターンを同時並行で解説していったので、流れが分かりにくくなっていたと思います。
パターンに分けてコードをまとめて記載するので、その通りやれば類似した結果が得られると思います。
Setting.txtの内容
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
####Caution#### All file names must not include space. Use "_" or "-" instead of space. ############### # Setting values about all scripts. Email = 'Your e-mail adress' Api_key = 'Your API key' # ==== Setting values about Download ID === ## Variables for efetch Database1 = nucleotide # Select the database to use. Rettype1 = fasta # Select the format to download files. Retmode1 = text # Select the file extension to download data. Count_in_query1 = 20 # How many queries to request at a time. Setting range is 1 to 100000 at a time. minday = 2000/01/01 Species = Plecoglossus altivelis Genetic = mitochondrion Feature = rRNA |
2-1. 対象IDの配列を遺伝子領域ごとにFastaファイルに出力するスクリプト
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 |
# --- import module --- # import subprocess as sp import pandas as pd import pathlib, glob, re, pprint, os, datetime, shutil, time, sys from Bio import Entrez, SeqIO # from Bio.GenBank.Record import Record from Bio.SeqFeature import SeqFeature, FeatureLocation from Bio.SeqRecord import SeqRecord from datetime import timedelta from time import sleep from dateutil.relativedelta import relativedelta from subprocess import Popen, PIPE from tqdm import tqdm from termcolor import colored, cprint # --- フォルダーのチェック --- # def check_results_folder(): # Reusltsフォルダの有無のチェック if pathlib.Path("./Results").is_dir() == True: shutil.rmtree("./Results") # フォルダを消して pathlib.Path("./Results").mkdir(exist_ok=True) # 作成 else: pathlib.Path("./Results").mkdir(exist_ok=True) # IDをまとめるファイルのチェック if pathlib.Path('./Results/ID.txt').is_file == True: pathlib.Path("./Results/ID.txt").unlink() else: with open('./Results/ID.txt', mode='w') as i: i.write("") # Fastaファイルをまとめるファイルのチェック if pathlib.Path("./Results/Fasta").is_dir() == False: pathlib.Path("./Results/Fasta").mkdir(exist_ok=True) else: pass # --- チェック --- # check_results_folder() # --- Color setting --- # class Color: BLACK = '\033[30m' RED = '\033[31m' GREEN = '\033[32m' YELLOW = '\033[33m' BLUE = '\033[34m' PURPLE = '\033[35m' CYAN = '\033[36m' WHITE = '\033[37m' END = '\033[0m' BOLD = '\038[1m' UNDERLINE = '\033[4m' INVISIBLE = '\033[08m' REVERCE = '\033[07m' # --- setting.txtの読み込み --- # with open('Setting.txt', encoding = "utf-8") as st: txt = st.read() # --- NCBIへの自己紹介 --- # Entrez.email = re.search(r'Email\s*=\s*(\S+)', txt).group(1) Entrez.api_key = re.search(r'Api_key\s*=\s*(\S+)', txt).group(1) # --- setting.txtから変数の情報を取得する --- # # データベースを取得 db = re.search(r'Database1\s*=\s*(\S+)', txt).group(1) # ファイルをダウンロードするときの形式 rettype = re.search(r'Rettype1\s*=\s*(\S+)', txt).group(1) # データをダウンロードするときのフォーマット retmode = re.search(r'Retmode1\s*=\s*(\S+)', txt).group(1) # 1回の要求でダウンロードするデータ数 count_in_query = re.search(r'Count_in_query1\s*=\s*(\S+)', txt).group(1) # いつからのデータを取得するか minday = re.search(r'minday\s*=\s*(\S+)', txt).group(1) # いつまでのデータを取得するか (常に今日になるようになっています) maxday = datetime.date.today().strftime("%Y/%m/%d") # 対象種を取得 species = re.search(r'Species\s*=\s*(.*)', txt).group(1) # 対象遺伝子を設定 genetic= re.search(r'Genetic\s*=\s*(\S+)', txt).group(1) # --- IDを検索する --- # handle_search = Entrez.esearch(db = db, retmax = count_in_query, term = f'{species}[Organism] AND {genetic}[filter]') record_search = Entrez.read(handle_search) # 取得したIDのList SeqID = record_search["IdList"] # 操作しやすいようにID毎に改行する str_ID = '\n'.join(SeqID) # ResultsフォルダのID.txtに取得したIDを書き出す with open('./Results/ID.txt', mode = 'a') as f: f.write(str_ID + "\n") # Dataframe の形でIDの読み込み ID_dataframe = pd.read_csv("./Results/ID.txt", encoding="UTF-8", header=None) # New function define def makeSeqRecord(seq_obj, id_for_fasta): record = SeqRecord(seq_obj, id=id_for_fasta, description="") return record # rRNA or CDS or tRNA misc_feature feature_str= re.search(r'Feature\s*=\s*(\S+)', txt).group(1) # Genbank ID数を取得 id_num = len(ID_dataframe) # Genbank ID の数だけForで情報を取得する for i in tqdm(range(0, id_num), desc = Color.CYAN + 'Progress rate' + Color.END): # IDの配列情報をGenbank形式で取得 handle_info = Entrez.efetch(db = db, id = ID_dataframe.iloc[i,0], rettype = 'gb', retmode = 'text') for record_info in SeqIO.parse(handle_info, "genbank"): # gbを指定するとGenbank形式の情報を取得する # リスト格納 record_list = [] # 配列全体を格納 parent_sequence = record_info.seq # 学名を格納 organism = record_info.annotations['organism'] # Accession numberを格納 acc = record_info.id # 各領域ごとに配列を切り分けて配分 for feature in record_info.features: # 不明な遺伝子領域 misc_feature 以外の場合 if feature.type != 'misc_feature' and feature_str == feature.type: sequence_slice = feature.location.extract(parent_sequence) # 配列の末端取得 strand_int = feature.location.strand if strand_int == 1: strand = 'top' # 5'末端 elif strand_int == -1: strand = 'btm' # 3'末端 else: strand = 'cannotbe' # 配列の開始位置指定 start = feature.location.parts[0].start.position + 1 # 配列の終了位置指定 end = feature.location.parts[-1].end.position # 遺伝子領域の情報取得 if 'gene' in feature.qualifiers: gene = feature.qualifiers['gene'][0] else: gene = 'NA' # 転写産物?の情報取得 if 'product' in feature.qualifiers: product = feature.qualifiers['product'][0] else: product = 'NA' # Fasta形式にした時の情報部分(> ~~~ の部分)の作成 id_line = acc + '_' + organism + '_' + gene + '_' + product + '_' id_line += ' (' + str(start) + '..' + str(end) + ', strand=' + strand + ')' # レコードから取得した情報をFasta形式にする record_list = makeSeqRecord(sequence_slice, id_line) # Fastaファイルの出力 if feature_str != 'D-loop': fasta_file_name = './Results/Fasta/' + acc + f'_{product}_' + feature.type + '.fasta' else: fasta_file_name = './Results/Fasta/' + acc + f'_{feature_str}_test.fasta' # 一応0以上かチェック type_count = len(record_list) with open(fasta_file_name, mode = 'w') as ffn: if type_count > 0: SeqIO.write(record_list, ffn, 'fasta') else : ffn.write('No Annotation Found') # merge fasta file name mfa = f'./Results/Fasta/mergefa.fas' with open(mfa, mode = 'a') as mfa: if type_count > 0: SeqIO.write(record_list, mfa, 'fasta') else: pass with open("./Results/list.txt", mode = 'w') as spl: spl.write(organism + '\n') # 不明な遺伝子領域 misc_feature の場合(D-loopがここに含まれる場合があるため作成) elif feature_str == 'misc_feature' and feature_str == feature.type or feature_str == 'misic_feature' and re.search('(d|D)-(l|L)oop', feature.type) == True: # feature.qualifiers フィールドにD-loopの記載がある場合があるため if 'note' in feature.qualifiers: note = feature.qualifiers['note'][0] else: note = 'No Annotation' sequence_slice = feature.location.extract(parent_sequence) # 配列の末端 strand_int = feature.location.strand if strand_int == 1: strand = 'top' # 5'末端 elif strand_int == -1: strand = 'btm' # 3'末端 else: strand = 'cannotbe' # 配列の開始位置 start = feature.location.parts[0].start.position + 1 # 配列の終了位置 end = feature.location.parts[-1].end.position # 転写産物?の情報 product = 'NA' # Fasta形式にした時の情報部分の作成 id_line = acc + '_' + organism + '_' + note + '_' id_line += ' (' + str(start) + '..' + str(end) + ', strand=' + strand + ')' # レコードから取得した情報をFasta形式にする record_list = makeSeqRecord(sequence_slice, id_line) # Fastaファイルの出力 fasta_file_name = './Results/Fasta/' + acc +'_' + feature.type + '.fasta' type_count = len(record_list) with open(fasta_file_name, mode = 'w') as ffn: if type_count > 0: SeqIO.write(record_list, ffn, 'fasta') else : ffn.write('No Annotation Found') # merge fasta file name mfa = './Results/Fasta/mergefa.fas' with open(mfa, mode = 'a') as mfa: if type_count > 0: SeqIO.write(record_list, mfa, 'fasta') else: pass # リストに追加された種名を記入 add_organism_list = [] add_organism_list = add_organism_list.append(organism) print(add_organism_list) with open("./Results/list.txt", mode = 'w') as spl: spl.write(organism + '\n') else: pass |
2-2.DNA領域に構わず、対象IDの配列を一括取得する
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 |
# --- import module --- # import subprocess as sp import pandas as pd import pathlib, glob, re, pprint, os, datetime, shutil, time, sys from Bio import Entrez, SeqIO # from Bio.GenBank.Record import Record from Bio.SeqFeature import SeqFeature, FeatureLocation from Bio.SeqRecord import SeqRecord from datetime import timedelta from time import sleep from dateutil.relativedelta import relativedelta from subprocess import Popen, PIPE from tqdm import tqdm from termcolor import colored, cprint # --- フォルダーのチェック --- # def check_results_folder(): # Reusltsフォルダの有無のチェック if pathlib.Path("./Results").is_dir() == True: shutil.rmtree("./Results") # フォルダを消して pathlib.Path("./Results").mkdir(exist_ok=True) # 作成 else: pathlib.Path("./Results").mkdir(exist_ok=True) # IDをまとめるファイルのチェック if pathlib.Path('./Results/ID.txt').is_file == True: pathlib.Path("./Results/ID.txt").unlink() else: with open('./Results/ID.txt', mode='w') as i: i.write("") # Fastaファイルをまとめるファイルのチェック if pathlib.Path("./Results/Fasta").is_dir() == False: pathlib.Path("./Results/Fasta").mkdir(exist_ok=True) else: pass # --- チェック --- # check_results_folder() # --- Color setting --- # class Color: BLACK = '\033[30m' RED = '\033[31m' GREEN = '\033[32m' YELLOW = '\033[33m' BLUE = '\033[34m' PURPLE = '\033[35m' CYAN = '\033[36m' WHITE = '\033[37m' END = '\033[0m' BOLD = '\038[1m' UNDERLINE = '\033[4m' INVISIBLE = '\033[08m' REVERCE = '\033[07m' # --- setting.txtの読み込み --- # with open('Setting.txt', encoding = "utf-8") as st: txt = st.read() # --- NCBIへの自己紹介 --- # Entrez.email = re.search(r'Email\s*=\s*(\S+)', txt).group(1) Entrez.api_key = re.search(r'Api_key\s*=\s*(\S+)', txt).group(1) # --- setting.txtから変数の情報を取得する --- # # データベースを取得 db = re.search(r'Database1\s*=\s*(\S+)', txt).group(1) # ファイルをダウンロードするときの形式 rettype = re.search(r'Rettype1\s*=\s*(\S+)', txt).group(1) # データをダウンロードするときのフォーマット retmode = re.search(r'Retmode1\s*=\s*(\S+)', txt).group(1) # 1回の要求でダウンロードするデータ数 count_in_query = re.search(r'Count_in_query1\s*=\s*(\S+)', txt).group(1) # いつからのデータを取得するか minday = re.search(r'minday\s*=\s*(\S+)', txt).group(1) # いつまでのデータを取得するか (常に今日になるようになっています) maxday = datetime.date.today().strftime("%Y/%m/%d") # 対象種を取得 species = re.search(r'Species\s*=\s*(.*)', txt).group(1) # 対象遺伝子を設定 genetic= re.search(r'Genetic\s*=\s*(\S+)', txt).group(1) # --- IDを検索する --- # handle_search = Entrez.esearch(db = db, retmax = count_in_query, term = f'{species}[Organism] AND {genetic}[filter]') record_search = Entrez.read(handle_search) # 取得したIDのList SeqID = record_search["IdList"] # 操作しやすいようにID毎に改行する str_ID = '\n'.join(SeqID) # ResultsフォルダのID.txtに取得したIDを書き出す with open('./Results/ID.txt', mode = 'a') as f: f.write(str_ID + "\n") # Dataframe の形でIDの読み込み ID_dataframe = pd.read_csv("./Results/ID.txt", encoding="UTF-8", header=None) # New function define def makeSeqRecord(seq_obj, id_for_fasta): record = SeqRecord(seq_obj, id=id_for_fasta, description="") return record # IDが記載されているID.txtの読み込み ID_dataframe = pd.read_csv("./Results/ID.txt", encoding="UTF-8", header=None) # Genbank ID数を取得 id_num = len(ID_dataframe) # GWnbank ID の数だけForで情報を取得する for i in tqdm(range(0, id_num), desc = Color.CYAN + 'Progress rate' + Color.END): handle = Entrez.efetch(db = db, id = ID_dataframe.iloc[i,0], rettype = 'gb', retmode = 'text') for record_info in SeqIO.parse(handle, "genbank"): # gbを指定するとGenbank形式の情報を取得する # リスト格納 record_list = [] # 配列全体を格納 parent_sequence = record_info.seq # 学名を格納 organism = record_info.annotations['organism'] # Accession numberを格納 acc = record_info.id # 各領域ごとに配列を切り分けて配分 for feature in record_info.features: # 不明な遺伝子領域 misc_feature 以外の場合 if feature.type == 'source': sequence_slice = feature.location.extract(parent_sequence) # 配列の末端取得 strand_int = feature.location.strand if strand_int == 1: strand = 'top' # 5'末端 elif strand_int == -1: strand = 'btm' # 3'末端 else: strand = 'cannotbe' # 配列の開始位置指定 start = feature.location.parts[0].start.position + 1 # 配列の終了位置指定 end = feature.location.parts[-1].end.position # Fasta形式にした時の情報部分(> ~~~ の部分)の作成 id_line = acc + '_' + organism + '(' + str(start) + '..' + str(end) + ', strand=' + strand + ')' # レコードから取得した情報をFasta形式にする record_list = makeSeqRecord(parent_sequence, id_line) # アクセッションナンバー.fa のファスタファイルでファイル名を指定 fasta_file_name = './Results/Fasta/' + record_info.id + '.fasta' # 一応0以上かチェック type_count = len(record_list) # 書き出し with open(fasta_file_name, mode = 'w') as ffn: if type_count > 0: SeqIO.write(record_list, ffn, 'fasta') else : ffn.write('No Annotation Found') # merge fasta file name mfa = './Results/Fasta/mergefa.fas' with open(mfa, mode = 'a') as mfa: if type_count > 0: SeqIO.write(record_list, mfa, 'fasta') else: pass |
使用用途は何なのかと言われると難しい所ですが、誰かの仕事や研究の手助けとなれば幸いです。





